
The Challenges - and our approaches
My career goal has been to understand the molecular basis of disease. I focus on the sequelae of DNA variation impacting on human health in the context of environmental and other genetic stressors, in order to rapidly deliver benefit to patients.
Delivering numeric confidence to guide personalised medicine is demanding in the setting of individually unique genomes each harbouring multiple rare, high-impact DNA variants absent from genome wide association studies, and barely represented in tissue banks from genomically- unselected donors. My group devises and validates methodologies that enhance opportunities to interrogate contributions of rare variants to health and disease.
Developing Reverse Translation, and why

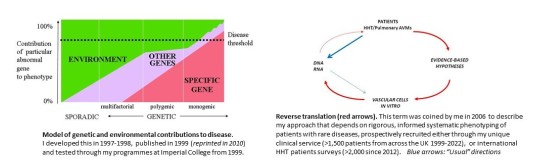
In 1997, based on my 1993-1996 observations in 36 families (>300 individuals) with hereditary haemorrhagic telangiectasia (HHT) or adenosine deaminase deficiency, and earlier training in classical genetics, I developed an overarching model of human disease (left hand figure). This predicted that where genetic diseases bring an unspecified pathology closer towards a detectable threshold, monogenic diseases might serve as high-risk models for identification of environmental and other genetic factors contributing to pathological states. To test, while continuing to use traditional genetic approaches (right hand figure, blue arrows) to identify genes which when mutated, cause human disease, I supplemented by an approach I termed Reverse Translation (right hand figure, red arrows). This required set up of dual clinical and research programmes that have taken 22 years and >1500 patients to come to ‘academic’ fruition, meanwhile delivering clinical benefit for more than 15 years.
The principal target has been a multisystemic inherited condition, hereditary haemorrhagic telangiectasia (HHT), that results from causative, pathogenic, loss-of-function “mutations” in genes that encode transforming growth factor (TGF)-β/bone morphogenetic protein (BMP) superfamily members. HHT variants cause two main vascular malformation types: (i) arteriovenous malformations (AVMs) that develop in utero/childhood, remain in adult life, and cause complications (most commonly strokes due to pulmonary AVMs), and (ii) adult onset, dynamic, but numerically progressive, smaller telangiectasia that are responsible, via bleeding/anaemia, for most of the day-to-day burden of disease and healthcare [Shovlin 2020]. HHT is clinically and mechanistically distinct from other inherited and vascular malformation syndromes, and has been my major focus because:
- The clinically important complications (strokes through right-to-left paradoxical emboli; iron deficiency anaemia) are relevant to more than one-third of the human population, transcending the rare disease origin;
- I was puzzled by the extreme clinical variability within members of the same family (from non-penetrance i.e. no clinical sequelae, to very severe [Shovlin, 1994]) and hypothesised that their study would illuminate modifiable risks relevant to the general population [Shovlin, 1999, reprinted in Shovlin, 2010];
- The patients provide a source of blood-derived cells to examine a molecular process operational in patient cells (nonsense mediated decay, Shovlin 1997) but obscured by conventional recombinant technological approaches.
Molecular causes- spectra of consequences
- My early contributions to HHT genetics (Shovlin 1994, Shovlin 1997) highlighted intrafamilial phenotypic diversity with the potential paucity of symptoms and signs further highlighted in 2022 (Anderson 2022) with data that changed the National Genomic Test Directory.
- Through the 100,000 Genomes Project we have (i) identified a new HHT disease gene (GDF2) (Balanchandar 2022); (ii) demonstrated that 10-15% mosaicism missed by Sanger is detectable by WGS (Clarke 2020), (iii) validated a novel filter developed by Sihao Xiao under my supervision, that reduces ~5 million variants per DNA to ~20,000 restricted to regions of functional significance (Xiao 2023), enabling machine learning to identify stress-associated variants in the 3'UTR; and (iv) applied the data to feedback to clinical diagnostic algorithms (Shovlin 2023).
- Through novel precision phenotyping of patients, supplementing usual binary phenotypic discriminators with validated severity scales, we were able to (i) demonstrate that secondary phenotypes (bleeding/anaemia) are not related to HHT causal genes (Shovlin 2020; Joyce 2022); (ii) show platelet/coagulation gene deleterious variants to be commonly present, and less well tolerated in HHT than the general population (Joyce 2022); (iii) develop a platform ‘towards personalised genomic interpretations’ (TPGI) (Joyce 2022) expected to be relevant to discrimination of phenotypes in other conditions, and (iv) use unsupervised machine learning algorithms to categorise HHT haematological indices into novel profiles that parallel distinct clinical haemorrhagic phenotypes (Mukhtar 2023). All are expected to be relevant to discrimination of phenotypes in other conditions.
Variability in same donor endothelial cells
Recognising that >60% of loss-of-function DNA variants lead to premature termination codons predicted to be degraded by nonsense mediated decay (NMD, Govani 2013), with evidence in HHT-derived EBV-transformed lymphoblastoid cells (Shovlin 1997), my group examined general consequences in endothelial cells (Bielowka 2023), and took forward by establishing blood outgrowth endothelial cells (BOECs) from pre-genotyped HHT patients. Our HHT BOEC transcriptome data is currently available as a preprint (Bernabeu-Herrero 2023), with a Qeios review regarding the implication for the specific rare disease. The data also identified a second, more generic process that explained in part, the variability between replicate cultures from the same donor, and related to differing degrees of NMD efficiency which could be quantified at 78-92% per culture.
Iron treatment - a phenotypic modifier
Multiple Shovlin 2016-2019 manuscripts demonstrate that iron treatments (prescribed >6 million times p.a. in England, and generators of reactive oxygen species (ROS)), are associated with endothelial injury(eg (Mollet 2016; Shovlin 2016a) and for a subgroup of HHTpatients, can aggrevate bleeding and anaemia (Shovlin 2016a; Shovlin 2016b; Shovlin 2016c; Boother 2017; Thielemans 2019). We are currently examining the cellular basis of this responses in our new test system resuspending peripheral blood mononuclear cells (PBMCs) in their own plasma (Xiao 2023) .
2020-2024 Genomic Publications
- Bernabéu-Herrero et al, Blood, Mar 2024
Mutations causing premature termination codons discriminate and generate cellular and clinical variability in HHT - Xiao et al, Am J Hum Genet Nov 2023
Functional filter for whole-genome sequencing data identifies HHT and stress-associated non-coding SMAD4 polyadenylation site variants >5 kb from coding DNA. - Shovlin et al, Br J Haematol. Jan 2024 MEK 1 inhibition and bleeding in hereditary haemorrhagic telangiectasia.
- Jain et al J Clin Med. Dec 2023
Pathogenic Variant Frequencies in Hereditary Haemorrhagic Telangiectasia Support Clinical Evidence of Protection from Myocardial Infarction. - McCarley et al J Clin Med. Dec 2023
Pharmacogenomic Considerations for Anticoagulant Prescription in Patients with Hereditary Haemorrhagic Telangiectasia. - Joyce et al Blood Advances, Jul 2022
Whole genome sequences discriminate hereditary hemorrhagic telangiectasia phenotypes by non-HHT deleterious DNA variants - Anderson et al Thorax, Jun 2022
Pulmonary arteriovenous malformations may be the only clinical criterion present in genetically confirmed hereditary haemorrhagic telangiectasia - Balanchander et al, Am J Med Genet Mar 2022
Identification and validation of a novel pathogenic variant in GDF2 (BMP9) responsible for hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations - Shovlin et al, J Med Genet, Aug 2023
Updates on diagnostic criteria for hereditary haemorrhagic telangiectasia in the light of whole genome sequencing of 'gene-negative' individuals recruited to the 100 000 Genomes Project - Clarke et al J Med Genet, Dec 2020
Low grade mosaicism in hereditary haemorrhagic telangiectasia identified by bidirectional whole genome sequencing reads through the 100,000 Genomes Project clinical diagnostic pipeline - Shovlin et al Blood, Oct 2020
Mutational and phenotypic characterization of hereditary hemorrhagic telangiectasia
2021-2023 HHT/PAVM Clinical Manuscripts
For hereditary haemorrhagic telangiectasia (HHT), the 2022 Frameworks manuscript from the European Reference Network for HHT (VASCERN HHT) summarises all of the 100% consensus statements generated 2016-2020 while chaired by Shovlin, and expands on key underlying circulatory physiology principles required to understand and appropriately manage HHT:
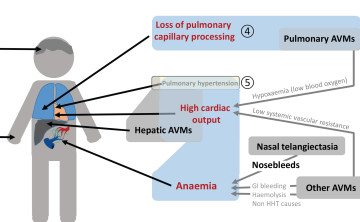 Anaemia, bleeding and iron intake
Anaemia, bleeding and iron intake- Pulmonary capillary bypass
Pulmonary AVMs - High cardiac output
Low systemic vascular resistance (AVMs), anaemia, hypoxaemia

The Curaçao Criteria (clinical diagnostic criteria developed in 1998 by the Shovlin-led Committee) have been readdressed following new insights from the 100,000 Genomes Project.

For pulmonary arteriovenous malformations (PAVMs), the 2017 Clinical Statement from the British Thoracic Society, chaired by Shovlin, remains valid.
We generated further data and insights into the mechanisms of exercise capacity/dyspnoea, and ischaemic stroke, summarised in the 2021 Orphanet Statement. Particular attention is drawn to our 2022 ischaemic stroke review article in Neurology (Topiwala, 2022).
2022/3 Selected Research Presentations
[1] AoPGBI Association of Physicians
Dublin,10-11 March 2022
New paradigms for “truncated proteins” in medicine and biology: Failure to degrade associated with reduced expression of injury-responsive RNA transcripts
[2] Genomics England Research Summit
London, 4 May 2022
Deciphering the rare and impactful: Development and application of functional tools for rare variant interrogation in genomic medicine
[3] Biochemical Society
Sheffield, 30 June 2022
Understanding protein consequences of inefficient co-translational nonsense mediated decay – Insights from RNASeq of patient-derived endothelial cells
[4] Gordon Research Conference on Post Transcriptional Gene Regulation Maine, 10-15 July 2022 (poster)
Towards an understanding of generic consequences from inefficient nonsense mediated decay.
[5] AoPGBI Association of Physicians
Liverpool, 20-21 April 2023
Loss-of-Function, or "Loss-of-Function Plus" PTC variant? Patients and their cells support a new classification system explaining phenotypic and treatment variability
[6] American Society for Human Genetics
Washington DC, 1-5 November 2023 (Reviewers' Choice Abstract)
Functional filter for whole genome sequencing data identifies ultrarare SMAD4 polyadenylation site variants >5kb from coding DNA in patients with hereditary hemorrhagic telangiectasia
Guest Lectures
Achievement Award for Rare Pulmonary Disease Lecture, European Respiratory Society, Vienna, Austria, 2012
Presidential Lecture, American Society of Haematology, New Orleans, US, 1999
>65 invited international and national lectures, including, American Thoracic Society; International Society on Thrombosis and Haemostasis; Royal Colleges of Physicians (London and Edinburgh); Royal Society of Medicine; British Thoracic Society (including two scientific plenary lectures); and multiple academic institutions in, US, Canada, Japan, Argentina, Spain, Germany, Italy, Turkey and UK, from, 1993