
Research Overview
DREAMLAB: harnessing the idle processing power of tens of thousands of smartphones, network-based AI and large -omics data to discover bioactive components of foods (“hyperfoods”) and novel indications of marketed drugs against cancer genomes and emerging COVID-19 disease. This is one of the largest citizen science project on AI-driven interrogation of “-omics“ data in precision medicine/nutrition with over ~250K engaged users. The solution was featured by technology programs such as BBC Click/Sky Swipe. The discoveries underpin the design of next-generation cancer preventative/therapeutic nutrition strategies.


- Laponogov I and Gonzales G, et al Veselkov K (2021) Network machine learning maps phytochemically rich “Hyperfoods” to fight COVID-19. Human Genomics 15, 1.
- Veselkov K*, et al, HyperFoods: Machine intelligent mapping of cancer-beating molecules in foods (2019), Scientific Reports, 9, 9237. Journal Top 25 Collection.
BASIS: BIOINFORMATICS PLATFORM FOR PROCESSING LARGE-SCALE MASS SPECTROMETRY IMAGING IN CHEMICALLY AUGMENTED HISTOLOGY
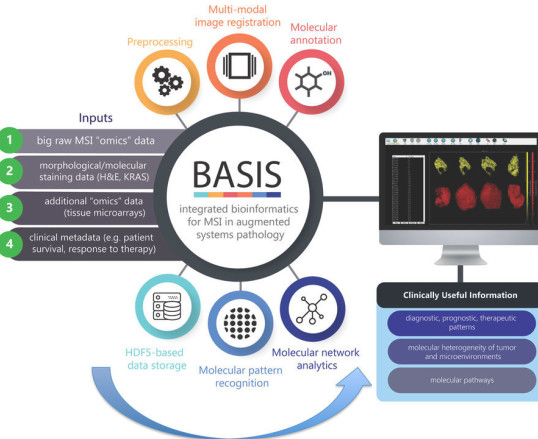
BASIS: High-performance and open-source machine learning platform for processing of large-scale Mass Spectrometry Imaging (MSI) data in chemically augmented histology Within healthcare, stratified medicine and digital pathology remains underexploited. MSI has the potential to deliver a paradigm shift in digital pathology services and cancer diagnostics by augmentation of cellular morphological analysis with in-depth data on cellular molecular content. In the process, a vast quantity of unrefined data (that can amount to several hundred gigabytes per tissue section) is produced. I have led the development of a field-changing data platform (“BASIS”) capable of optimized and scalable machine-learning processing of MSI data from hundreds to thousands of samples. From importing raw mass spectrometry data to integration with other data forms (morphological data, clinical metadata) to optimized pre-processing, image analysis, molecular annotation, and pattern learning, “BASIS” transforms raw MSI data into clinical insights. This BASIS platform has been applied for MSI datasets to discriminate lymph node metastasis in gastric cancer, 3D molecular imaging, heterogeneity assessment and deep molecular interrogation of various cancers.
Veselkov KA*, et al (2018) BASIS: High-performance bioinformatics platform for processing of large-scale mass spectrometry imaging data in chemically augmented histology, Scientific Reports, 8, 4053.
Veselkov KA*,et al (2014). Chemo-informatic strategy for imaging mass spectrometry-based hyperspectral profiling of lipid signatures in colorectal cancer, PNAS, 111: 1216-122.
Abbassi-Ghadi N, Antonowicz S, McKenzie J,Kumar S, Huang J, Jones E, Strittmatter N, Petts G, Kudo H, court S, Hoare J, Veselkov K, Goldin R, Takats Z, Hanna G, (2020) De novo lipogenesis alters the phospholipidome of esophageal adenocarcinoma, Cancer Research, 80: 2764-2774.
Abbassi-Ghadi N, Golf O, Kumar S, Antonowicz S, McKenzie JS, Huang J, Strittmatter N, Kudo H, Jones EA, Veselkov K, Goldin R, Takáts Z, Hanna GB (2016) Imaging of esophageal lymph node metastases by desorption electrospray ionization mass spectrometry, Cancer Research.
MSHUB AND CHEMDISTILLER: PROCESSING, ANNOTATION AND INTERROGATION OF LARGE-SCALE CHROMATOGRAPHY-MASS SPECTROMETRY DATA
We have identified a massive bottleneck in the existing processing, annotation and data analytics platforms available for chromatography-mass spectrometry data and tandem MS data. Currently, existing solutions do not scale well with large datasets (>1000 samples) and therefore rendering large impactful metabolic projects unfeasible.My team members (Drs Ivan Laponogov, Dennis Veselkov) have thus developed a highly scalable server-based chromatography–mass spectrometry processing platform, named “MSHub”. The platform uses latest advances in computational algorithms and machine-learning methods, as well as a complimentary user-friendly frontend that enables the tool to be used by non-experts. The unique features of the MSHub platform are analytical transparencyand reproducibility, workflow versatility, and scalability.

The output of the MSHub pre-processing workflow includes fragmentation spectra and quantitative integrals of molecules. The fragmentation spectra require reliable annotations. We have thus introduced ChemDistiller, a customizable engine that combines automated large-scale annotation of metabolites using tandem MS data with a compiled database containing tens of millions of compounds with pre-calculated ‘fingerprints’ and fragmentation patterns. Our tests using publicly and commercially available tandem MS spectra for reference compounds show retrievals rates comparable to or exceeding the ones obtainable by the current state-of-the-art solutions in the field while offering higher throughput, scalability and processing speed.

Aksenov AA, Laponogov I, et al Veselkov K* (2020) Auto-deconvolution and molecular networking of gas chromatography–mass spectrometry data. Nature Biotechnology.
Laponogov I, Sadawi N, Dieter G, Mirnezami R, Veselkov K* (2018) ChemDistiller: an engine for metabolite annotation in mass spectrometry, Bioinformatics, 34(12): 2096-2102.
HASKEE: Hypothesis and AssociationS from KnowledgE and Evidence natural language processing platform
‘Omics’-data-driven studies often report and summarize findings in terms of putative molecular markers or pathways effected. However, few of these are further investigated or reach clinical practice, resulting in a waste of resources and research. While validation should be performed as a first step by systematic review and meta-analysis of literature, the increase in annual publications has rendered this impossible to perform manually – taking a collaborator over a year to perform literature review of chemotherapy response studies in colorectal cancer and summarized in Figure.
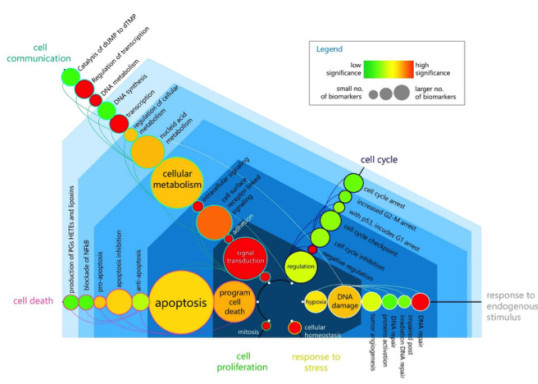
To mitigate and facilitate this, we have developed a natural language processing pipeline and tool (HASKEE) to automate this process. HASKEE (hypothesis and associations from knowledge and evidence) is a search engine that allows users to search for evidence supporting a hypothesis, or to explore and infer potential biological associations that have not been reported before. As part of the project, we have benchmarked existing solutions and identified a generalizability issue, which we report in our manuscript. Improving on existing solutions, we currently have a working viable pipeline, with the full open source code and fully-evaluated tool aimed to be released in the next year.
Galea D, Laponogov I, Veselkov K* (2018), Exploiting and assessing multi-source textual data for supervised biomedical named entity recognition, Bioinformatics, 34(14):2474-2482.
Collaborators
Dr Tania Fleitas Kanonnikoff, INCLIVA, Artificially Intelligence Diagnostic Assistant for Gastric Inflammation, 2021
Professor Vasilis Vasiliou, Yale University, AI/Machine Learning for multi-omics and network-structured data, 2019
Professor Michael Bronstein, University of Oxford, Network AI/Graph Deep Learning, 2016
Prof George Hanna, Division of Surgery, Imperial College London, MSHub: Bioinformatics platform for processing and annotation of large-scale chromatography-mass spectrometry data, 2016
Prof Pieter Dorrestein and Dr Alexander Aksenov, University of San Diego, Processing and annotation of large-scale mass spectrometry data, 2014
Prof Jeremy Everett, University of Greenwich, Computational modelling for dynamic pharaco-metabonomics, 2013
Guest Lectures
BASIS: Open-source and high-performance bioinformatics platform for mass spectrometry imaging in chemically augmented histology, School of Public Health, Yale University, USA, 2017
Optimized processing workflow for improved information recovery from large-scale MSI data, 65th ASMS Conference, June 4-8, 2017, 2017
Translational Bioinformatics for Mass Spectrometry in Systems Medicine, International workshop on Translational Bioinformatics and Health Informatics, Izmir, Turkey, 2015
Data analytics for mass spectrometry in stratified medicine, OurCon III: Imaging Mass Spectrometry Conference 2015, Piza, Italy, 2015
Top-down systems biology approaches for “omics”-based tissue and biofluid analytics, Proceedings of CELLmicrocosmos neXt 2014, Bielefeld, Germany, 2014
Enhanced tissue-specific molecular pattern extraction via recursive maximum margin criterion, 3-rd International Conference on Machine Learning and Computer Science, http://psrcentre.org/images/extraimages/2%20114011.pdf, 2014
Novel data processing and image co-registration algorithm for region-specific lipid profiling in colorectal cancer tissue using DESI imaging mass spectrometry, The Association of Surgeons of Great Britain and Ireland (ASGBI)’s 2013 International Surgical Congress; Nominated for the Association’s prestigious Moynihan Prize., Glasgow, UK, 2013
Translational bioinformatic platform for imaging mass spectrometry, BMMS annual meeting 2013, Eastbourne, UK., 2013
The iKnife: Analysis of Diathermy Plumes by High Resolution Mass Spectrometry provides Real Time Identification of Colorectal Cancer Liver Metastases, 6th London Surgical Symposium, Imperial College London. Best presentation award., Imperial College London, UK, 2012
Metabolic entropy approach for measuring systemic disruptions in patho-physiological states, Harvard Medical School, Boston, 2006
Enhanced tissue-specific molecular pattern extraction via recursive maximum margin criterion, 3-rd International Conference on Machine Learning and Computer Science, Dubai, United Arab Emirates