
Summary
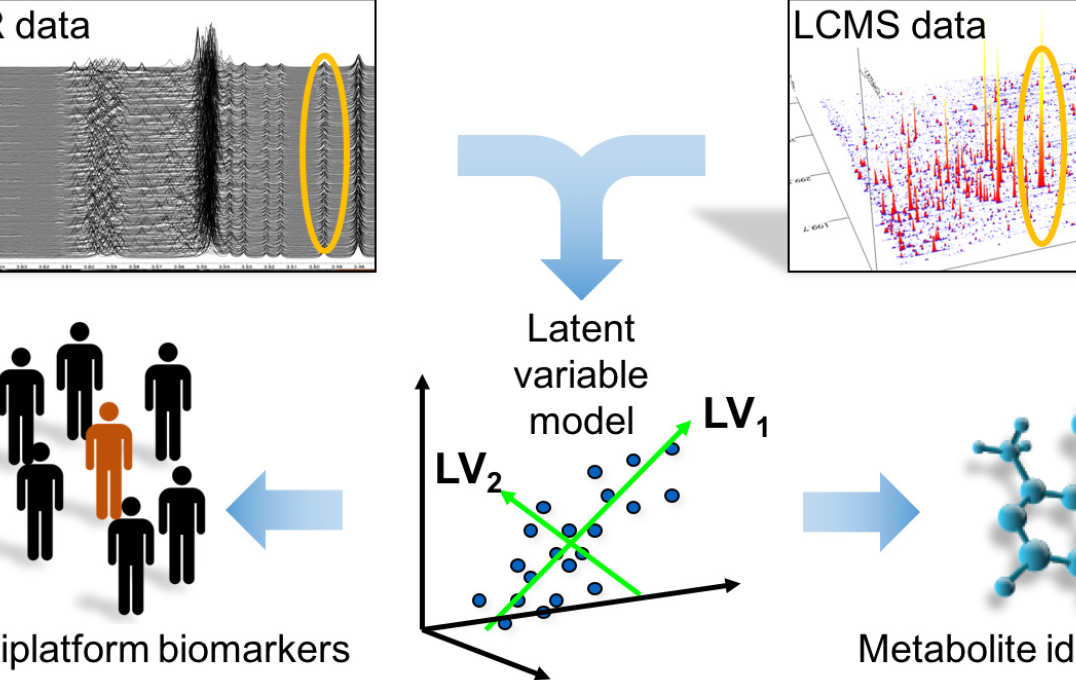
Metabolomic data integration
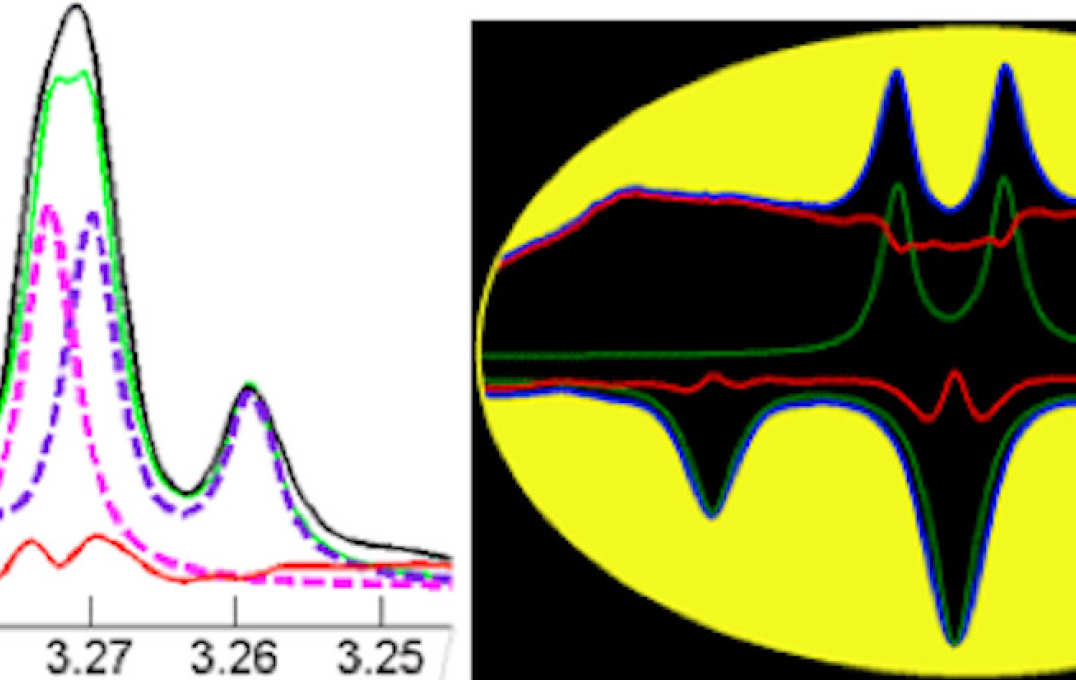
BATMAN NMR modelling
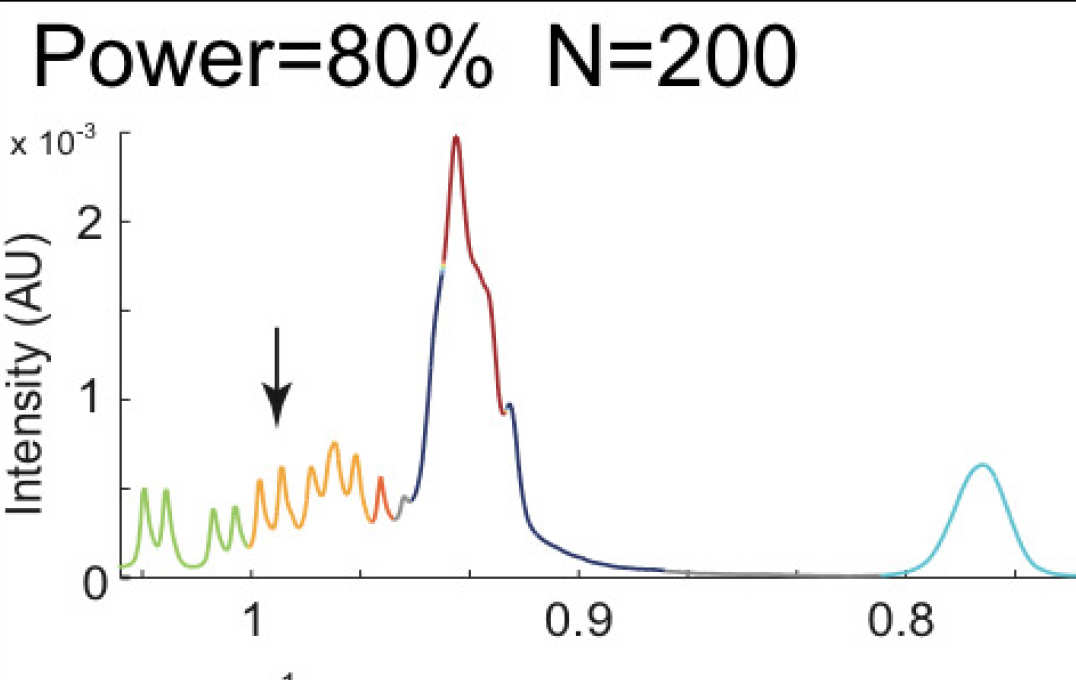
Metabolomics power analysis
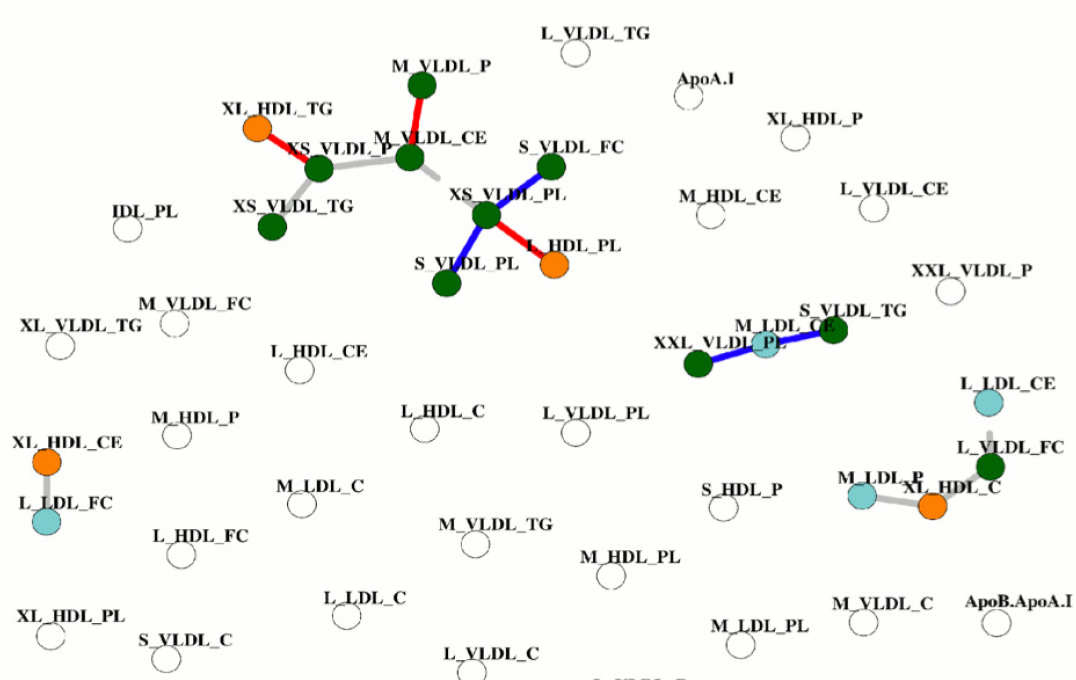
Differential association networks
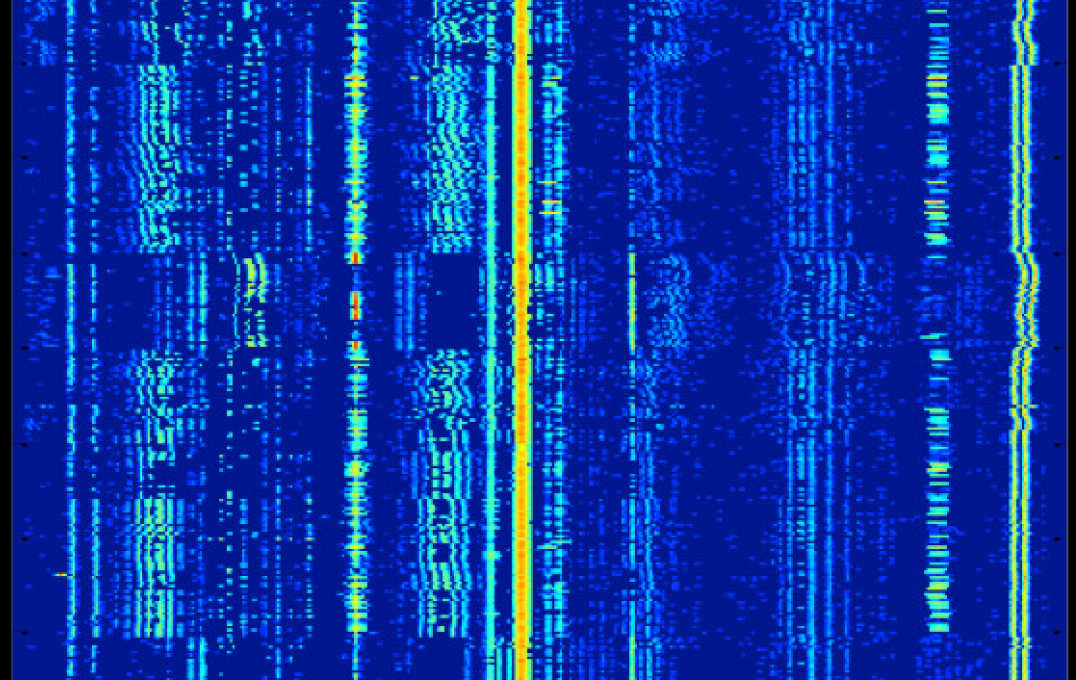
Large scale metabolomic data processing
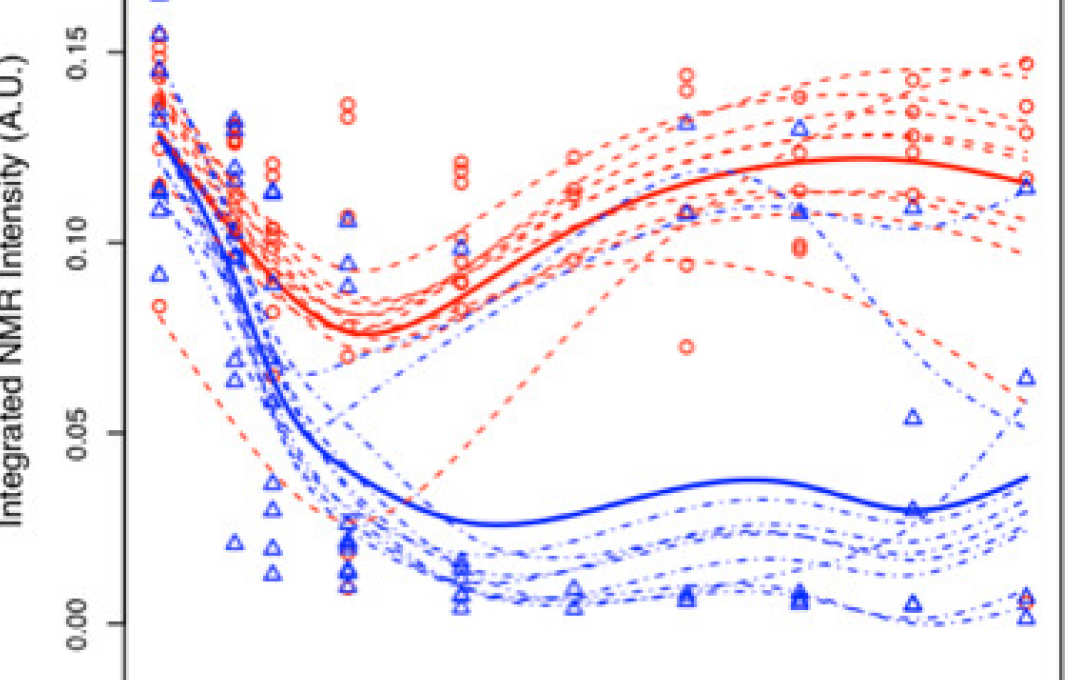
Short time series analysis
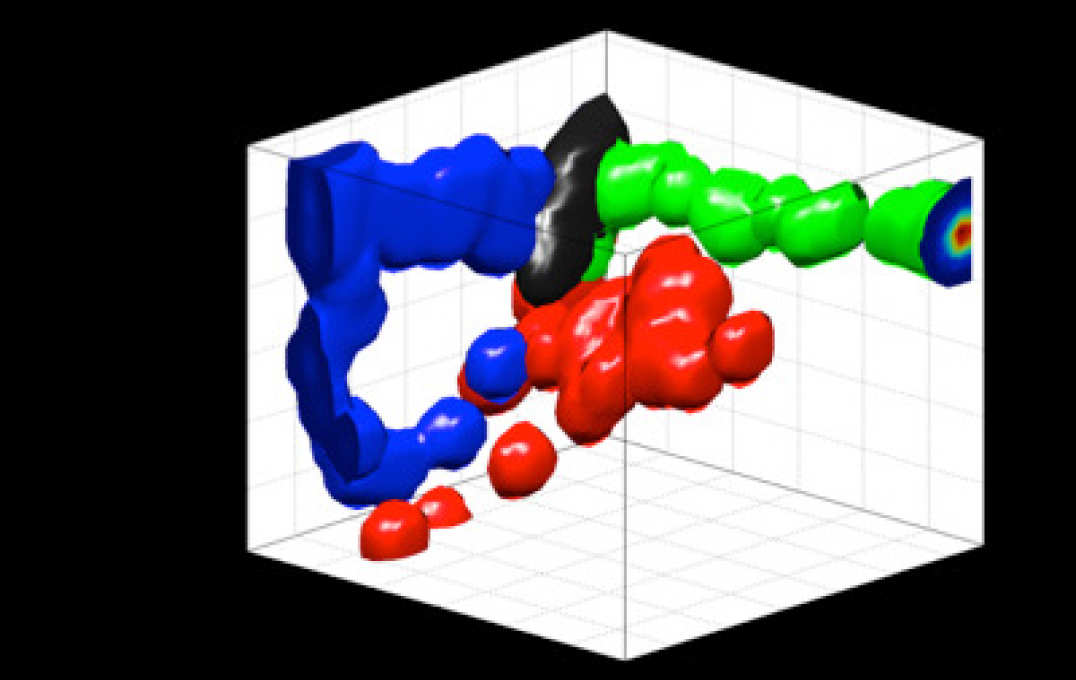
Data visualisation
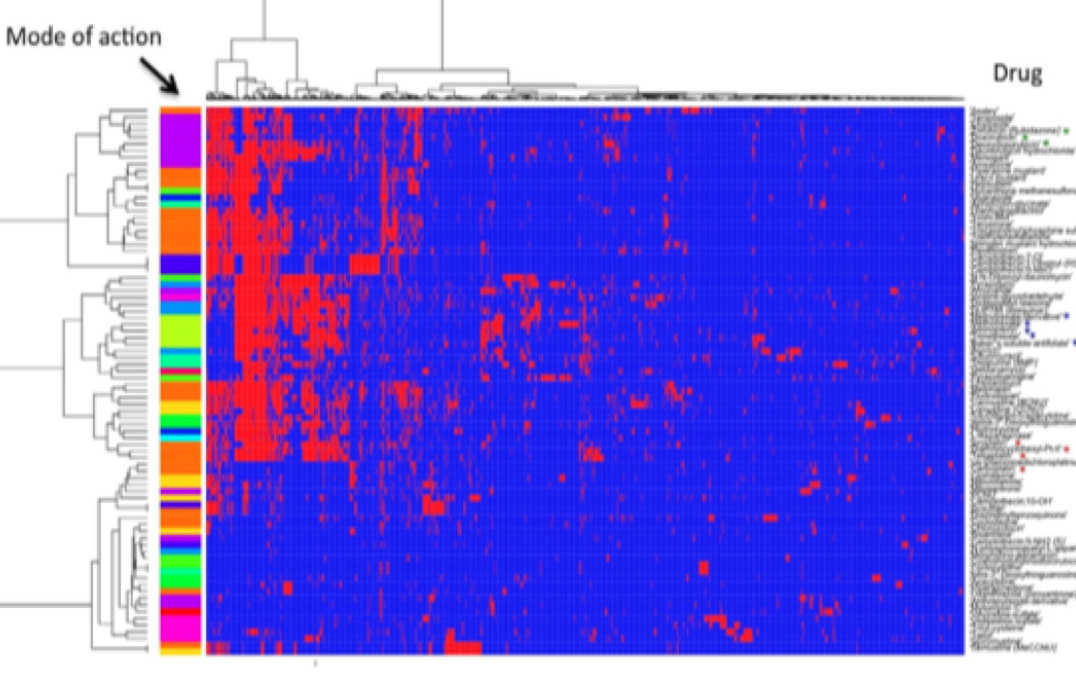
Metabolomic/transcriptomic pathway analysis
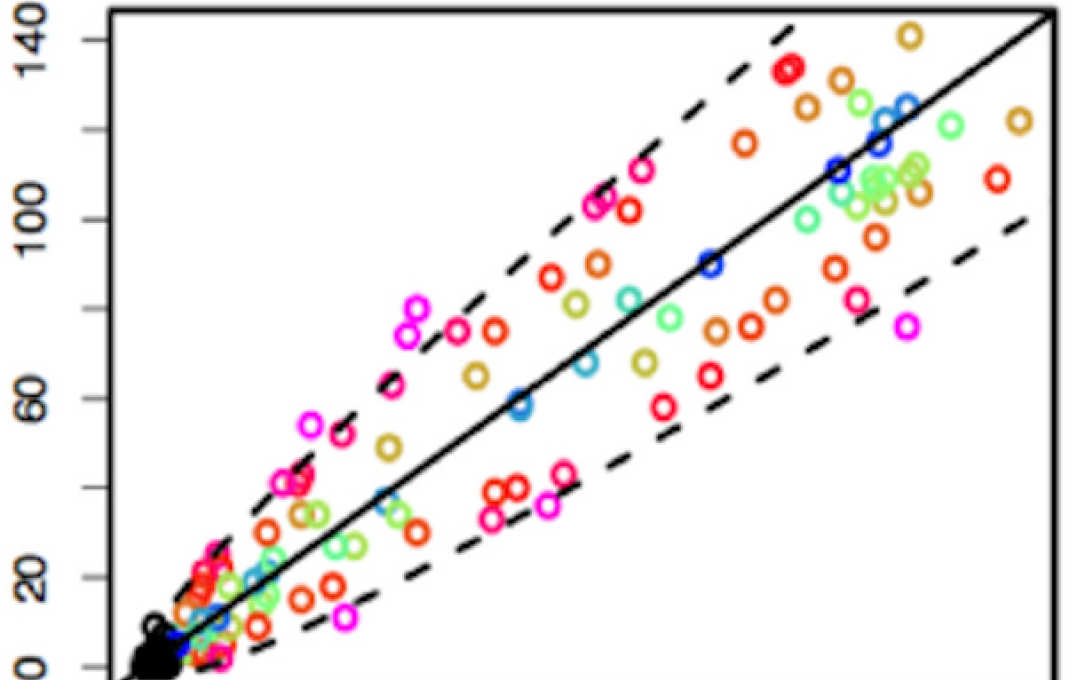
Modelling mass spectrometry data
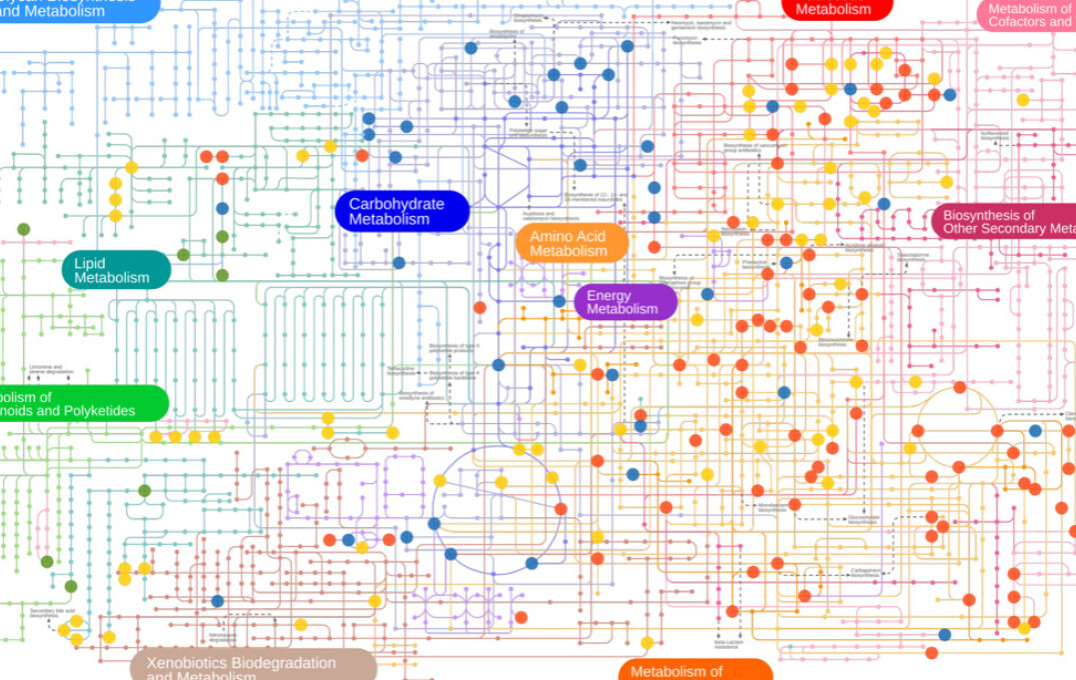
Metabolic networks and pathways
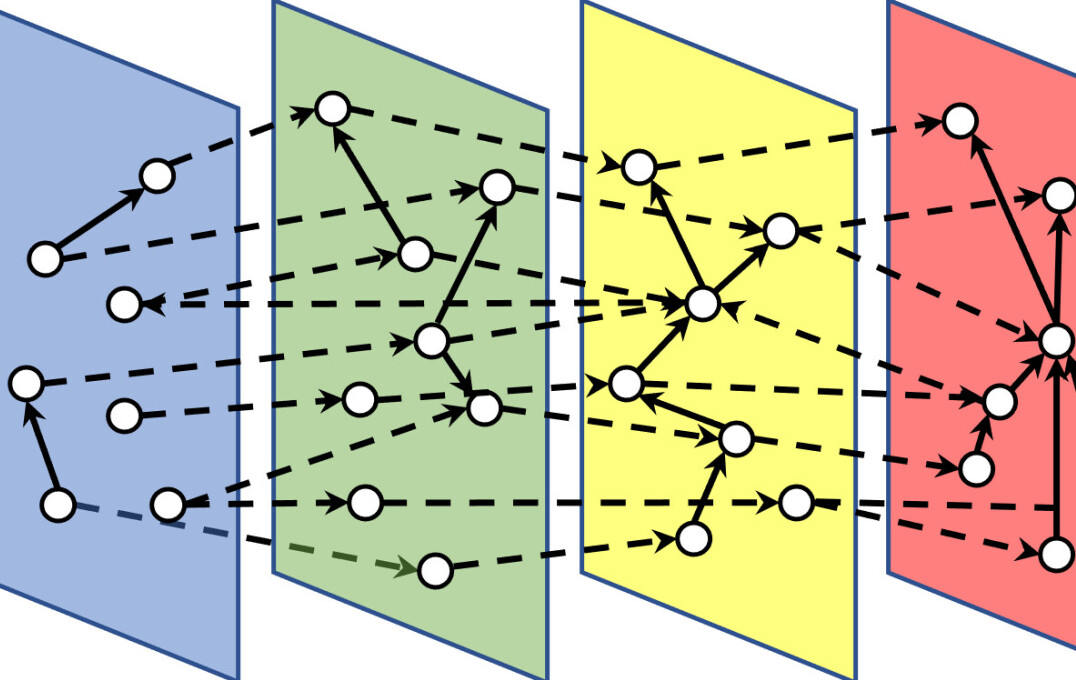
Multi-omics integration











I am Professor of Biomedical Data Science and Head of the Section of Bioinformatics within the Division of Systems Medicine of the Department of Metabolism, Digestion and Reproduction. I am also Director of the MRes in Biomedical Research and co-lead for its Data Science stream. My overall research interests lie at the interface between two broad areas:
multivariate data analysis,
and
post-genomic technologies.
More specifically, on the computational side these include, machine learning, artificial intelligence, bioinformatics, chemometrics, and multivariate statistics, and on the experimental side, the fields of genomics, transcriptomics and metabolomics. I am interested in applying diverse computational and mathematical methods in order to disentangle the mass of information at multiple biological levels generated by the –omics technologies. The ultimate aim is to synthesise the information provided by each of these techniques, thus facilitating a multi-scale understanding of biological systems. My main area of interest is computational metabolomics: solving the problems of metabolomics through computational and statistical means. These broad aims lead to several themes in my research:
- Improving information extraction from Nuclear Magnetic Resonance (NMR) spectroscopy & Liquid Chromatography–Mass Spectrometry (LC-MS) metabolic profiles
- Novel methods for predictive modelling of post-genomic data
- Statistical association networks as complex phenotypes in post-genomics
- Statistical integration and visualisation of metabolic profiles with other post-genomic data
- Time series analysis of post-genomic data
- Computational identification and annotation of metabolomics data.
Current projects are detailed on my Research page.
teaching

Dr Tim Ebbels introduces the MRes in Biomedical Research
Selected Publications
Journal Articles
Tzoulaki I, Castagné R, Boulangé CL, et al., 2019, Serum metabolic signatures of coronary and carotid atherosclerosis and subsequent cardiovascular disease, European Heart Journal, Vol:40, ISSN:1522-9645, Pages:2883-2896