


The dynamics of methylammonium ions in hybrid organic–inorganic perovskite solar cells
Over the past few years hybrid organic-inorganic perovskites have shown great promise as solar absorbers in photovoltaic cells. Solar cells based on these materials have enjoyed meteoric rises in efficiency in a relatively short space of time, however there remains several unanswered questions concerning their physics and operation.
One of these questions arises when we consider the behaviour of these devices under different applied voltage (bias) conditions. The photocurrent in these devices has been seen to be dependent on the previous bias conditions, or in other words the current voltage properties show a hysteresis.
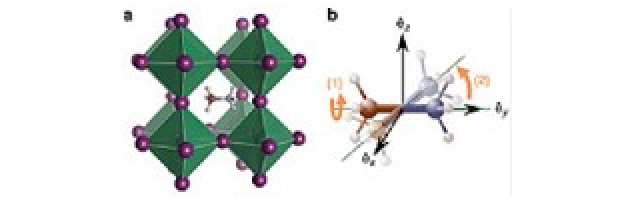 The origin of this behaviour is currently unknown, but one hypothesis is that the organic components (methylammonium ions in the case of the hybrid perovskite studied here), which act as rotationally mobile electrostatic dipoles, may rearrange themselves in an ordered fashion similar to dipoles in ferroelectric or anti-ferroelectric materials, which are also hysteretic.
The origin of this behaviour is currently unknown, but one hypothesis is that the organic components (methylammonium ions in the case of the hybrid perovskite studied here), which act as rotationally mobile electrostatic dipoles, may rearrange themselves in an ordered fashion similar to dipoles in ferroelectric or anti-ferroelectric materials, which are also hysteretic.
In order to study how the organic molecule in this material behaves, ab initio molecular dynamics calculations and neutron scattering measurements were first performed. This showed that the organic molecules like to 'hop' between different discrete orientations inside the cage created by the inorganic components (see Figure). These results also showed that the majority of the methylammonium ions were actually not rotationally mobile over a wide temperature range. We hypothesised that this could be due to long range ordering of the dipoles into 'stable domains', which would act to greatly suppress the rotation of the ions.
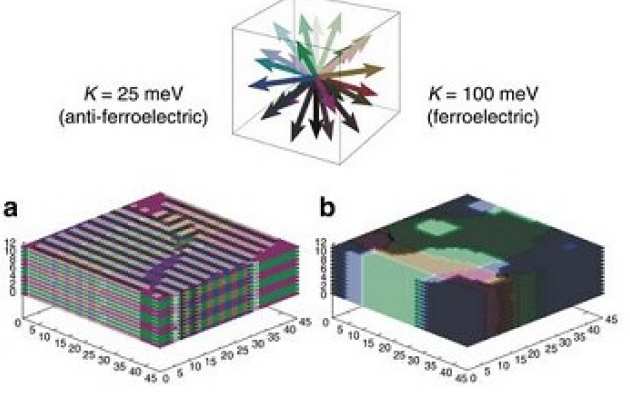 Andrew McMahon (TSM-CDT Cohort 5) then performed a series of Monte Carlo simulations in order to test this 'domain' hypothesis. The simulations were set up in order to 'freeze-in' some domain structures and calculate a defined measure of the probability of rotation throughout the simulation cell. This did indeed show, assuming only electrostatic interactions between the ions, that domains should greatly suppress rotation, with rotation far more likely to occur at the boundaries between domains ('domain walls'). By calculating the change in probability of rotation for the ions after applying an electric field in the simulation, a timescale associated with hysteresis could be derived. This was shown to most likely be too short to be consistent with observed experimental hysteresis.
Andrew McMahon (TSM-CDT Cohort 5) then performed a series of Monte Carlo simulations in order to test this 'domain' hypothesis. The simulations were set up in order to 'freeze-in' some domain structures and calculate a defined measure of the probability of rotation throughout the simulation cell. This did indeed show, assuming only electrostatic interactions between the ions, that domains should greatly suppress rotation, with rotation far more likely to occur at the boundaries between domains ('domain walls'). By calculating the change in probability of rotation for the ions after applying an electric field in the simulation, a timescale associated with hysteresis could be derived. This was shown to most likely be too short to be consistent with observed experimental hysteresis.
These results provide an important addition to the literature and will hopefully stimulate further discussion concerning the role of the organic component in these materials and the role it plays in observed photovoltaic device behaviour.
This work has just been published in Nature Communications.