All investigators associated with the CSB can be shown in a list or sorted by research theme or by technique.
Research highlights
γ-proteobacteria eject their polar flagella under nutrient depletion, retaining flagellar motor relic structures.
The Beeby lab recently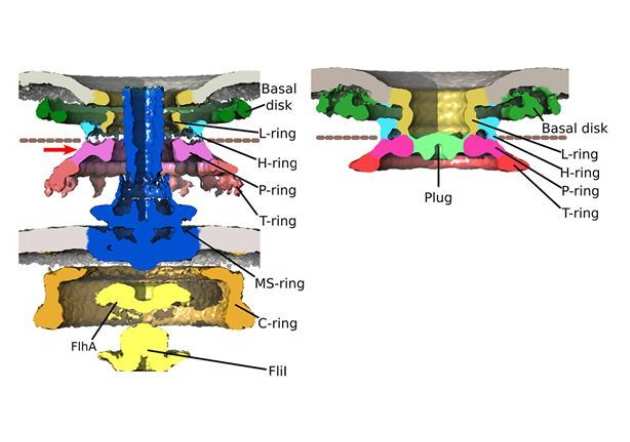 used high-throughput electron cryo-tomography to discover the unexpected finding that some bacteria take the drastic measure of ejecting their flagella in response to nutrient deficiency. Bacteria continually assemble flagella as propellers—unrelated to eukaryotic flagella—rotated by rotary motors embedded in the cell; continual rotation and assembly can consume up to 3% of a bacterium’s energy. Using electron cryo-tomography, a technique that provides high-resolution 3D images of intact bacteria, we were surprised to find partial flagellar motors in bacterial cells that were rare when nutrients were abundant but became common when nutrients were scarce. A variety of clues led us to hypothesize that these structures were relics of motors whose flagella had been ejected, which we confirmed using a genetic approach. Curiously, flagellar relics—which would otherwise be open portals through which the contents of the bacterial periplasm could leak—were plugged by an unidentified protein, presumably as a preservation measure. We speculated that flagellar ejection saves the bacterium from the costs of continuously assembling and rotating its flagella, as a last-ditch survival attempt. Our work provided a striking example of evolution arriving at a functional yet unintuitive solution to a problem, and the power of electron cryo-tomography to bridge molecular and cellular scales (Ferreira et al. PLOS Biology 2019).
used high-throughput electron cryo-tomography to discover the unexpected finding that some bacteria take the drastic measure of ejecting their flagella in response to nutrient deficiency. Bacteria continually assemble flagella as propellers—unrelated to eukaryotic flagella—rotated by rotary motors embedded in the cell; continual rotation and assembly can consume up to 3% of a bacterium’s energy. Using electron cryo-tomography, a technique that provides high-resolution 3D images of intact bacteria, we were surprised to find partial flagellar motors in bacterial cells that were rare when nutrients were abundant but became common when nutrients were scarce. A variety of clues led us to hypothesize that these structures were relics of motors whose flagella had been ejected, which we confirmed using a genetic approach. Curiously, flagellar relics—which would otherwise be open portals through which the contents of the bacterial periplasm could leak—were plugged by an unidentified protein, presumably as a preservation measure. We speculated that flagellar ejection saves the bacterium from the costs of continuously assembling and rotating its flagella, as a last-ditch survival attempt. Our work provided a striking example of evolution arriving at a functional yet unintuitive solution to a problem, and the power of electron cryo-tomography to bridge molecular and cellular scales (Ferreira et al. PLOS Biology 2019).
DNA translocation mechanism of an XPD family helicase
DinG (DNA damage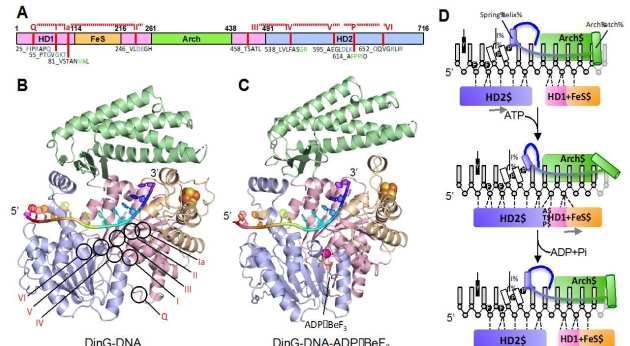 -inducible protein G in E. coli) belongs to the Xeroderma pigmentosum D (XPD) family. It is a Superfamily 2 DNA helicase with 5’−3’ directionality. DinG comprises four domains (panel A) the helicase (HD1 and HD2), arch and FeS cluster domains. Crystal structures of complexes of DinG bound to single-stranded DNA (ssDNA) were determined in the absence (panel B) and presence (panel C) of an ATP analogue (ADP•BeF3). Binding of nucleotide induces a conformational change that involves closure of a cleft between the two canonical helicase domains. Concomitant changes in the interactions between the protein and the bound ssDNA result in unidirectional translocation in a 5’−3’ direction (panel D). The structures improve our understanding of the translocation mechanism of XPD family enzymes as well as explaining how some of the mutations in these proteins cause deficiencies in their activities that have consequences in human disease (Cheng et al. eLife 2018).
-inducible protein G in E. coli) belongs to the Xeroderma pigmentosum D (XPD) family. It is a Superfamily 2 DNA helicase with 5’−3’ directionality. DinG comprises four domains (panel A) the helicase (HD1 and HD2), arch and FeS cluster domains. Crystal structures of complexes of DinG bound to single-stranded DNA (ssDNA) were determined in the absence (panel B) and presence (panel C) of an ATP analogue (ADP•BeF3). Binding of nucleotide induces a conformational change that involves closure of a cleft between the two canonical helicase domains. Concomitant changes in the interactions between the protein and the bound ssDNA result in unidirectional translocation in a 5’−3’ direction (panel D). The structures improve our understanding of the translocation mechanism of XPD family enzymes as well as explaining how some of the mutations in these proteins cause deficiencies in their activities that have consequences in human disease (Cheng et al. eLife 2018).
Structure and single molecule analysis of the SWR1 chromatin remodeller reveal details of ATP-dependent histone exchange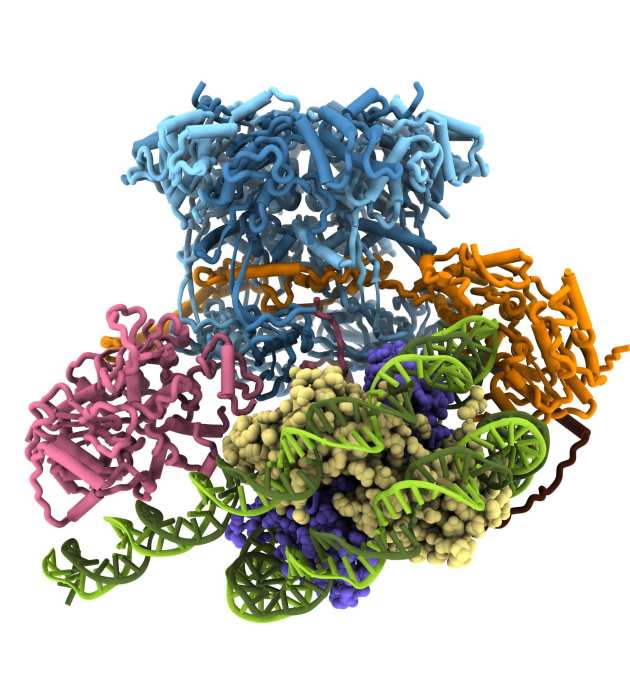
Eukaryotic chromatin is made up of nucleoprotein complexes known as nucleosomes, which act as signalling hubs for vital cellular activities. The protein component of these nucleosomes comprises a core of eight histone proteins, of which there are many subtypes that influence different events. The 14-subunit SWR1 chromatin remodelling complex from yeast catalyses the ATP-dependent exchange of canonical H2A histones for the Htz variant, which is an important signal in DNA damage repair and transcription. Members of the Wigley lab captured the initial collision complex of SWR1 with its nucleosome substrate and solved its structure at 3.6 Å using cryo-electron microscopy. In collaboration with the Rueda lab, the group were able to further characterise the enzyme complex using single-molecule fluorescence techniques. In short, SWR1 induces three major changes to the nucleosome: translocation of the nucleosomal DNA, flexing of the histone core and significant unwrapping of the nucleosomal entry DNA. These results highlight the preliminary distortions required to carry out histone exchange, and provides the first insight into the mechanistic basis for a poorly understood process (Willhoft et al. Science 2018)
Structures of Bacterial RNA Polymerase Complexes Reveal the Mechanism of DNA Loading and Transcription Initiation
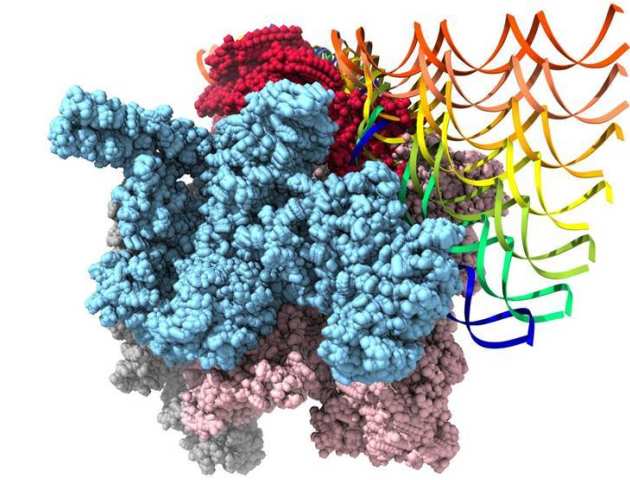 Gene transcription is carried out by multi-subunit RNA polymerases (RNAPs). Transcription initiation is a dynamic multi-step process that involves the opening of the double-stranded DNA to form a transcription bubble and delivery of the template strand deep into the RNAP for RNA synthesis. Applying cryoelectron microscopy to a unique transcription system using σ54 (σN), the major bacterial variant sigma factor, Professor Xiaodong Zhang's group capture a new intermediate state at 4.1 Å where promoter DNA is caught at the entrance of the RNAP cleft. Combining with new structures of the open promoter complex and an initial de novo transcribing complex at 3.4 and 3.7 Å, respectively, their studies reveal the dynamics of DNA loading and mechanism of transcription bubble stabilization that involves coordinated, large-scale conformational changes of the universally conserved features within RNAP and DNA. In addition, their studies reveal a novel mechanism of strand separation by σ54 (Glyde et al.. Mol Cell 2018).
Gene transcription is carried out by multi-subunit RNA polymerases (RNAPs). Transcription initiation is a dynamic multi-step process that involves the opening of the double-stranded DNA to form a transcription bubble and delivery of the template strand deep into the RNAP for RNA synthesis. Applying cryoelectron microscopy to a unique transcription system using σ54 (σN), the major bacterial variant sigma factor, Professor Xiaodong Zhang's group capture a new intermediate state at 4.1 Å where promoter DNA is caught at the entrance of the RNAP cleft. Combining with new structures of the open promoter complex and an initial de novo transcribing complex at 3.4 and 3.7 Å, respectively, their studies reveal the dynamics of DNA loading and mechanism of transcription bubble stabilization that involves coordinated, large-scale conformational changes of the universally conserved features within RNAP and DNA. In addition, their studies reveal a novel mechanism of strand separation by σ54 (Glyde et al.. Mol Cell 2018).
Fragment-derived inhibitors of human N-myristoyltransferase block capsid assembly and replication of the common cold virus
Rhinoviruses are the most frequent cause of the common cold and the most frequent cause of exacerbation in asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis, resulting in billions of dollars in healthcare costs in the US alone. There is currently no specific treatment for rhinovirus infections, since the diversity of viral serotypes greatly impedes clinical efficacy of virus-targeted therapies or vaccines. The alternative paradigm of targeting a host process on which the virus depends is very attractive, since the host is an invariant factor in the infection. In Mousnier et al., Nature Chemistry 2018 (in press) we demonstrate for the first time that the human enzymes that modify rhinovirus capsid with a myristate fatty acid (NMT1 and NMT2), are a viable pharmacological target in rhinovirus infection.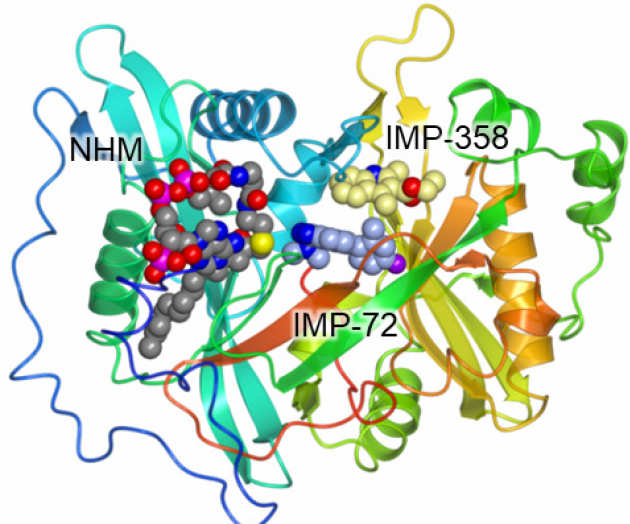
We describe a fragment redesign and merging approach (Fig. 1) which allowed us to obtain an exceptionally potent human NMT inhibitor, IMP-1088. X-ray structures for this novel series in complex with human NMT1 reveal the binding modes underpinning our optimisation approach and explain the drug-like potency of IMP-1088, which is the first example of a picomolar IC50 dual human NMT1/2 inhibitor. The selectivity of IMP-1088 was confirmed through chemical proteomic analyses of myristoylation in cells infected with rhinovirus, which additionally provide the first direct proteomic evidence for N-myristoylation of rhinovirus capsid in infected host cells.
We go on to present multiple lines of biological and chemical evidence showing that NMT inhibition potently blocks intact capsid formation without affecting viral genome or protein production, resulting in complete inhibition of virus replication. This novel mode of action is effective across multiple rhinovirus strains, in a primary human epithelial infection model, and against other important pathogens of the same family (polio and foot-and-mouth disease viruses), indicating the broad potential of this host-targeted strategy. Furthermore, IMP-1088 presents no cytotoxicity over the course of multiple rounds of infection (Mousnier et al. Nature Chem 2018).
Crystal structure of the sialic acid transporter SiaT in an outward-open conformation.
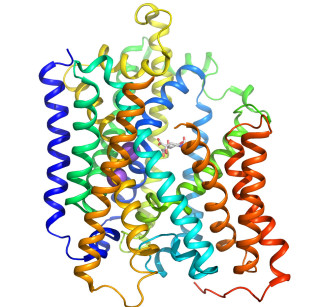 Many pathogenic bacteria utilise sialic acids as an energy source or use them as an external coating to evade immune detection. As such, bacteria that colonise sialylated environments deploy specific transporters to mediate import of scavenged sialic acids. Here, we report a substrate-bound 1.95 Å resolution structure and subsequent characterisation of SiaT, a sialic acid transporter from Proteus mirabilis. SiaT is a secondary active transporter of the sodium solute symporter (SSS) family, which use Na+ gradients to drive the uptake of extracellular substrates. SiaT adopts the LeuT-fold and is in an outward-open conformation in complex with the sialic acid N-acetylneuraminic acid and two Na+ ions. One Na+ binds to the conserved Na2 site, while the second Na+ binds to a new position, termed Na3, which is conserved in many SSS family members. Functional and molecular dynamics studies validate the substrate-binding site and demonstrate that both Na+ sites regulate N-acetylneuraminic acid transport.
Many pathogenic bacteria utilise sialic acids as an energy source or use them as an external coating to evade immune detection. As such, bacteria that colonise sialylated environments deploy specific transporters to mediate import of scavenged sialic acids. Here, we report a substrate-bound 1.95 Å resolution structure and subsequent characterisation of SiaT, a sialic acid transporter from Proteus mirabilis. SiaT is a secondary active transporter of the sodium solute symporter (SSS) family, which use Na+ gradients to drive the uptake of extracellular substrates. SiaT adopts the LeuT-fold and is in an outward-open conformation in complex with the sialic acid N-acetylneuraminic acid and two Na+ ions. One Na+ binds to the conserved Na2 site, while the second Na+ binds to a new position, termed Na3, which is conserved in many SSS family members. Functional and molecular dynamics studies validate the substrate-binding site and demonstrate that both Na+ sites regulate N-acetylneuraminic acid transport.
Structure and regulation of the human INO80–nucleosome complex
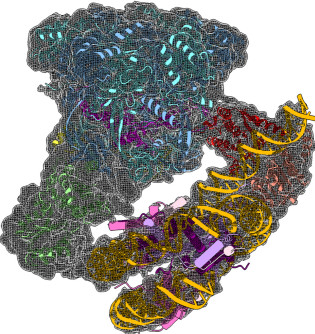
Using cryoEM, the collaborative work from the Wigley and Zhang labs reveals how the multi-subunit chromatin remodeller INO80 engages with a nucleosome and how the interactions induce distortions in the nucleosome, in preparation for the remodelling. The structures reveals a unique nucleosome-interaction mode involving two major contact points, located on opposite sides of the nucleosome (superhelical location -2 and superhelical location -6), in contrast with other known chromatin remodellers which mainly contact nucleosome through one common interaction site (superhelical location +2) (Ayala et al. Nature 2018).
High-Throughput Kinetic Analysis for Target-Directed Covalent Ligand Discovery
Cysteine-reactive small molecules are used as chemical probes of biological systems and as medicines. Identifying high-quality covalent ligands requires comprehensive kinetic analysis to distinguish selective binders from pan-reactive compounds. He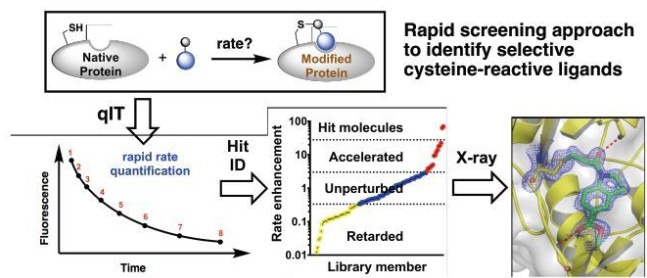 re we describe quantitative irreversible tethering (qIT), a general method for screening cysteine-reactive small molecules based upon the maximization of kinetic selectivity. We apply this method prospectively to discover covalent fragments that target the clinically important cell cycle regulator Cdk2. Crystal structures of the inhibitor complexes validate the approach and guide further optimization. The power of this technique is highlighted by the identification of a Cdk2-selective allosteric (type IV) kinase inhibitor whose novel mode-of-action could be exploited therapeutically (Craven et al. Angew Chem Int Ed Engl. 2018)
re we describe quantitative irreversible tethering (qIT), a general method for screening cysteine-reactive small molecules based upon the maximization of kinetic selectivity. We apply this method prospectively to discover covalent fragments that target the clinically important cell cycle regulator Cdk2. Crystal structures of the inhibitor complexes validate the approach and guide further optimization. The power of this technique is highlighted by the identification of a Cdk2-selective allosteric (type IV) kinase inhibitor whose novel mode-of-action could be exploited therapeutically (Craven et al. Angew Chem Int Ed Engl. 2018)
Electron cryo-tomography reveals evolution of high-power bacterial flagellar motors
 Dr Morgan Beeby's group used electron cryo-tomography to visualize bacterial flagellar motors in situ to probe how different bacteria swim with different abilities. The structures provided a quantitative explanation: bacteria that are better able to swim in higher viscosities can do so having evolved considerably larger motors with wider rings of additional motor proteins. Subsequently the group has collected data to propose a first model for how these motors evolved.
Dr Morgan Beeby's group used electron cryo-tomography to visualize bacterial flagellar motors in situ to probe how different bacteria swim with different abilities. The structures provided a quantitative explanation: bacteria that are better able to swim in higher viscosities can do so having evolved considerably larger motors with wider rings of additional motor proteins. Subsequently the group has collected data to propose a first model for how these motors evolved.
PNAS 113 (13) E1917-E1926 (2016) | Imperial website
Scientific Reports volume 8, Article number: 97 (2018) | Imperial website
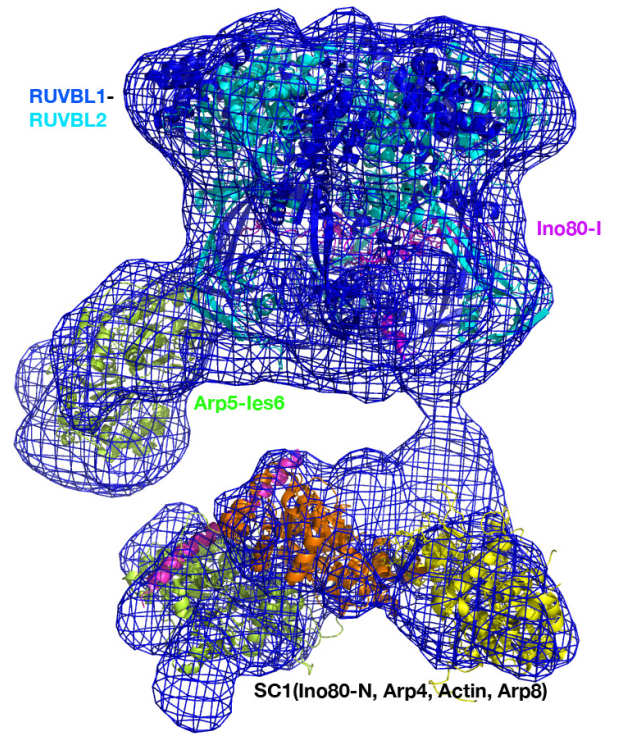
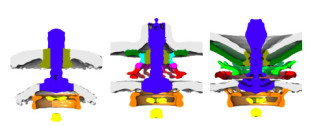
CryoEM structures of the human INO80 chromatin remodelling complex
Aramayo RJ, Willhoft O, Ayala R, Bythell-Douglas, R, Wigley DB, Zhang X (2018). Nat Struct Mol Biol. 25(1):37-44
The collaborative work between the Wigley and Zhang groups uses cryoEM to produce the first high resolution structure of a multi-subunit chromatin remodeller revealing the subunit arrangement of the INO80 chromatin remodeller as well as the structure and functional roles of key subunits within the complex.
Structural basis for antibacterial peptide self-immunity by the bacterial ABC transporter McjD
The ABC transporter McjD exports the antibacterial peptide MccJ25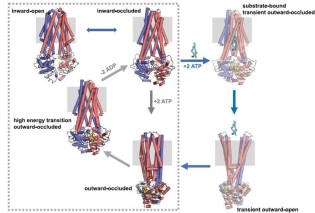 in Escherichia coli. we have determined its structure in a novel conformation, apo inward-occluded and a new nucleotide-bound state, high-energy outward-occluded intermediate state, with a defined ligand binding cavity. Predictive cysteine cross-linking in E. coli membranes and PELDOR measurements along the transportcycle indicate that McjD does not undergo major conformational changes as previously proposed for multi-drug ABC exporters. Combined with transport assays and molecular dynamics simulations, we propose a novel mechanism for toxic peptide ABC exporters that only requires the transient opening of the cavity for release of the peptide. We propose that shielding of the cavity ensures that the transporter is available to export the newly synthesized peptides, preventing toxic-level build-up (Bountra et al. EMBO J 2017)
in Escherichia coli. we have determined its structure in a novel conformation, apo inward-occluded and a new nucleotide-bound state, high-energy outward-occluded intermediate state, with a defined ligand binding cavity. Predictive cysteine cross-linking in E. coli membranes and PELDOR measurements along the transportcycle indicate that McjD does not undergo major conformational changes as previously proposed for multi-drug ABC exporters. Combined with transport assays and molecular dynamics simulations, we propose a novel mechanism for toxic peptide ABC exporters that only requires the transient opening of the cavity for release of the peptide. We propose that shielding of the cavity ensures that the transporter is available to export the newly synthesized peptides, preventing toxic-level build-up (Bountra et al. EMBO J 2017)
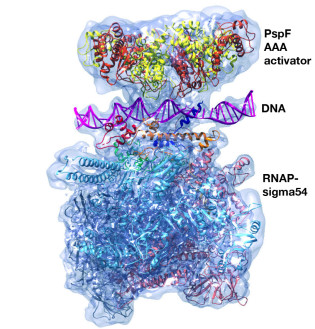
Structures of RNA Polymerase Closed and Intermediate Complexes Reveal Mechanisms of DNA Opening and Transcription Initiation
Glyde R, Ye F, Darbari VC, Zhang N, Buck M and Zhang X (2017). Mol Cell. 67(1):106-116
This collaborative cryoEM study betyween the Buck and Zhang groups provided the first structural information about a transcription intermediate complex and of a prokaryotic transcription closed promoter complex. The structures reveal how DNA opening is initiated during transcription.
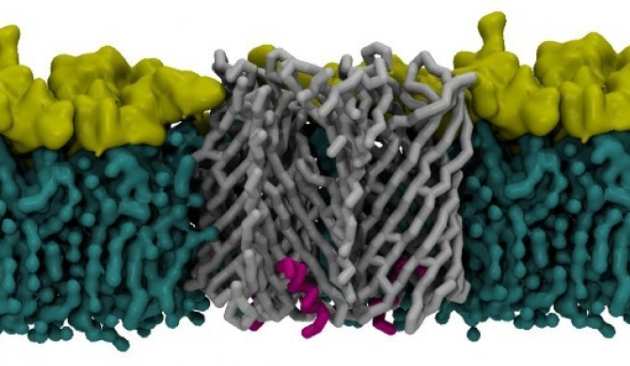
A new class of hybrid secretion system is employed in Pseudomonas amyloid biogenesis
Bacterial biofilms are a major cause of recurrent disease, allowing reservoirs of bacteria to persist in a human/animal host or in the external environment. A key component of these biofilms is functional amyloid - robust fibres that contribute to the mechanical properties of biofilm. Knowledge of the molecular properties of the proteins involved in amyloid formation is vital to our understanding of the means by which bacteria target hosts, evade the immune system, survive environmental stresses and cause disease. There are two genetically distinct functional amyloid systems are known in bacteria – the Curli (first characterised in E. coli) and Fap (Pseudomonas aeruginosa) extracellular fibre systems. Recent breakthroughs have led to new structural and mechanistic insight into curli biogenesis, but the Fap system is poorly understood. We present the high-resolution structure of the membrane protein transport component FapF showing that it belongs to a new family of multimeric β-barrel membrane pore. Combining NMR, X-ray and native MS data we reveal that FapF channel is trimeric and gated by a plug domain, which is regulated by a periplasmic coiled-coil. These observations are in contrast to those on the equivalent membrane component from the curli system, CsgG - an ungated nonomeric β-barrel pore. Remarkably, the Fap system does share some features with other outermembrane secretions systems. Mutagenesis together with biological assays and biophysical analysis support the structural view of how the FapF pore is activated by engagement with the pre-amyloid cargo and proteolysis. Our work unveils an alternative strategy of how gram negative bacteria transport amyloidogenic substrates across the outer membrane. Nature Communications, volume 8, Article number: 263 (2017)
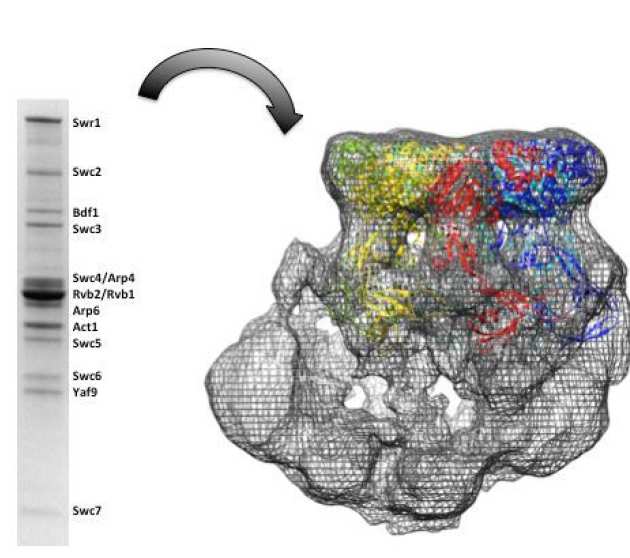
Characterization and architecture of yeast SWR1 histone exchange complex
Chromatin remodeling alters chromatin structure to make DNA more accessible for DNA regulatory proteins those are involved in the transcription, replication and DNA repair. SWR1 is an ATP-dependent chromatin remodeling complex that catalyzes the exchange of the variant histone Htz1 with the canonical histone H2A in the nucleosome. SWR1 complex is composed of fourteen subunits with a molecular weight about 1.07 MDa. We expressed and purified SWR1 complex from insect cells and the recombinant material is active in both gel and FRET-based histone exchange assays. We used subunit deletions and serial truncation of the Swr1 main subunit to investigate interactions between subunits of the SWR1 complex. Finally, we present electron microscopy studies revealing the dynamic nature of the complex and a 21 Å resolution reconstruction of the intact complex provides details not apparent in previously reported structures, including a large central cavity of sufficient size to accommodate a nucleosome (Lin et al. (2017) Nucleic Acids Res.).
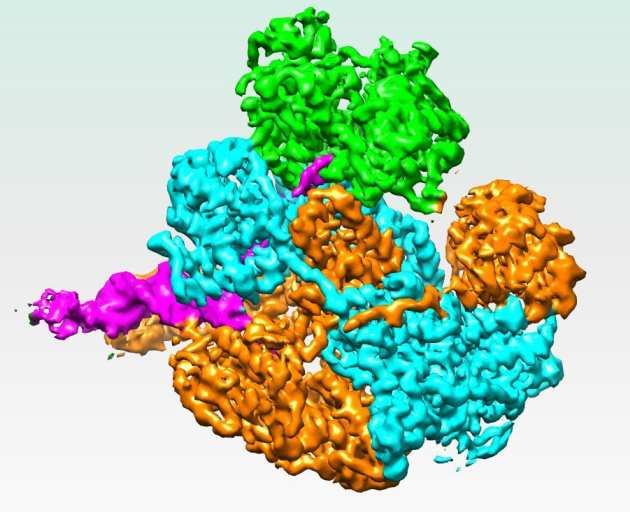
Mechanism for Nuclease Regulation in RecBCD
In Escherichia Coli, the helicase-nuclease protein complex RecBCD is responsible for processing double strand breaks for repair by homologous recombination. There is little structural information available about how the multiple activities of RecBCD are regulated. The group of Prof. Dale Wigley have solved a 3.8Å resolution cryo-EM structure of RecBCD with a DNA substrate that reveals a mechanism for nuclease activation within the complex. Interactions between the unwound 5’ ssDNA tail and RecD initiate conformational changes that transmit through all three subunits to release a blocking helix from the nuclease active site. A bacterial SH3-like domain plays a key role in the conformational changes that lead to activation (Wilkinson et al. (2016) eLife 5:e18227).

TssA forms a gp6‐like ring attached to the type VI secretion sheath
The type VI secretion system (T6SS) is a supra‐molecular bacterial complex that resembles phage tails. It is a killing machine which fires toxins into target cells upon contraction of its TssBC sheath. Here, we show that TssA1 is a T6SS component forming dodecameric ring structures whose dimensions match those of the TssBC sheath and which can accommodate the inner Hcp tube. The TssA1 ring complex binds the T6SS sheath and impacts its behaviour in vivo. In the phage, the first disc of the gp18 sheath sits on a baseplate wherein gp6 is a dodecameric ring. The Filloux and Freemont groups found remarkable sequence and structural similarities between TssA1 and gp6 C‐termini, and they propose that TssA1 could be a baseplate component of the T6SS. Furthermore, they identified similarities between TssK1 and gp8, the former interacting with TssA1 while the latter is found in the outer radius of the gp6 ring. These observations, combined with similarities between TssF and gp6N‐terminus or TssG and gp53, lead them to propose a comparative model between the phage baseplate and the T6SS (Planamente et al. (2016) EMBO J 35:1613).
 A Salmonella toxin promotes persister formation through tRNA acetylation
A Salmonella toxin promotes persister formation through tRNA acetylation
All bacterial species form persisters, which are cells that stop replicating and are tolerant to many antibiotics. Persisters are likely to be a cause of many recurrent infections, but little is known about how they arise. Salmonella, which causes gastroenteritis and typhoid fever, forms large numbers of non-replicating persisters after being engulfed by immune cells called macrophages. At least 14 Toxin-Antitoxin modules of Salmonella are involved in the formation of persisters after uptake by macrophages. Work from the Helaine and Hare groups led to the characterization of one of these toxins of a new type. They solved its crystal structure and revealed that it blocks protein production by acetylation of aminoacyl-tRNAs (Cheverton et al. (2016) Mol Cell 16:30139).

Functionalised carbon nanomaterials as nucleants for macromolecular crystallization
Naomi Chayen and colleagues report the first large-scale systematic analysis of a series of carbon nanomaterials (CNMs) with different surface functionalization for their effectiveness in inducing crystal nucleation. Functionalised CNMs are particularly attractive as their reactions tend to create combinations of surface area, porosity and surface heterogeneity (created by topography and chemistry) which are important for crystal nucleation of proteins and other macromolecules. Sparsely functionalized flat sheet geometries proved exceptionally effective leading to a new class of nucleant, based on PEG grafted graphene-related materials that can be applied to promote the growth of 3D crystals for X-ray crystallography (Govada et al. (2016)Scientific Reports Nature publishing Group 6:20053; Leese et al. (2016) Chemical Science 7, 2916-2923).
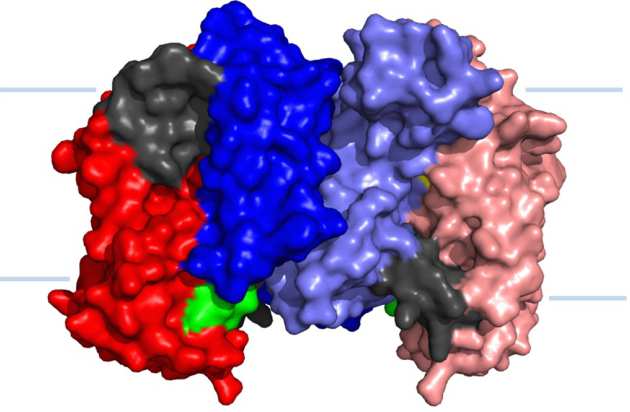
Structure of UapA suggest role for homodimerisation in transport activity
The high resolution structure of the eukaryotic purine/H+ symporter, UapA, in complex with xanthine revealed that the protein forms a closely associated dimer. The structure revealed novel details on the precise molecular basis substrate binding and selectivity and also showed that dimerisation plays a key role in transporter function (Alguel et al. 2016 Nature Communications 7:11336).