Computer Designed Molecules
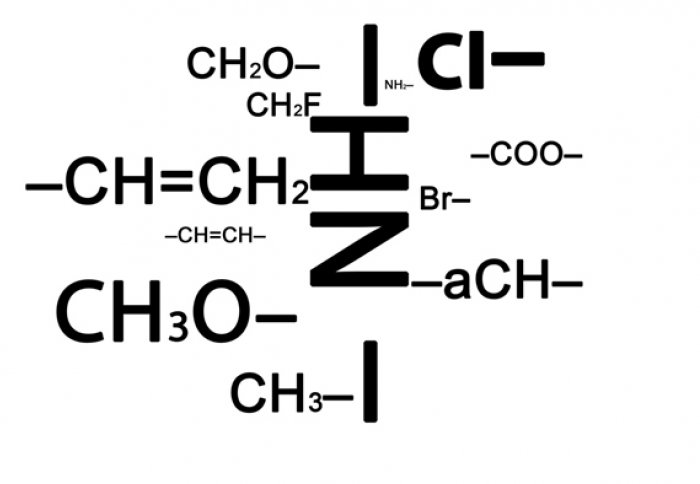
The solvent design space is made of building blocks that are used to construct the best molecule
Research published this week in Nature Chemistry outlines a novel methodology of selecting - and designing - solvents
Research published this week by Chemical Engineering researchers outlines a novel methodology for selecting solvents that could save industrial chemists a significant amount of time and money. The work, led by Professors Claire Adjiman and Amparo Galindo of the Molecular Systems Engineering (MSE) group, and Professor Alan Armstrong (Department of Chemistry), presents a computational method that allows engineers to optimise industrial processes by selecting, via a combination of quantum mechanical calculations and advanced optimisation techniques, the best choice of solvent for a given reaction from thousands of potential candidates. Traditionally, new candidates for use as a solvent would need to be tested experimentally prior to use, a process that is both costly and time-intensive; this approach can model with great accuracy how a solvent could slow or accelerate a given reaction without the need for an experimental approach.
The research, published in Nature Chemistry and carried out in collaboration with Syngenta, should prove very useful in the development of pharmaceutical or agrochemical manufacturing processes, where most reactions take place in a solvent and where the choice of reaction medium has many implications on downstream processing, affecting product quality, energy costs and environmental impact. According to Adjiman, “this publication is an exciting first step in the integrated optimisation of reactors, from molecules to process. Our method can be applied to a whole range of reactions, and can be used to take into account the many constraints that affect solvent choice.”
As well as being able to identify suitable molecules among common solvents, the method introduced by the research team goes even further by enabling the design of totally new solvents. Known as computer-aided molecular design (CAMD), the technique uses sophisticated algorithms to enable the identification of molecular structures that perform a chosen function the best way. The results can then be used to narrow down huge fields of possible molecular designs to just a handful of candidates which can then be verified experimentally. In the Nature Chemistry article, a design space of some 1,341 molecules was explored by CAMD in the application of the method to the well-known Menschutkin reaction, and a solvent was uncovered that is capable of increasing the rate of the reaction by 40%.
Article supporters
Article text (excluding photos or graphics) © Imperial College London.
Photos and graphics subject to third party copyright used with permission or © Imperial College London.
Reporter
Press Office
Communications and Public Affairs
- Email: press.office@imperial.ac.uk
