Crucial step in formation of deadly brain diseases discovered
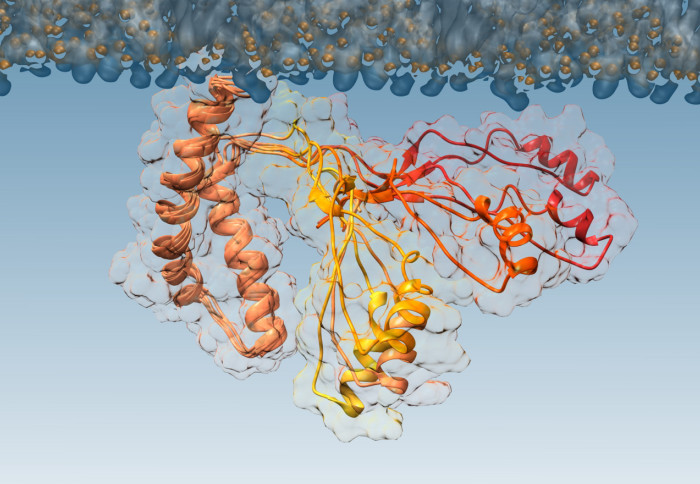
For the first time, researchers have pinpointed what causes normal proteins to convert to a diseased form, causing conditions like CJD and Kuru.
The research team, from Imperial College London and the University of Zurich, also tested a way to block the process, which could lead to new drugs for combatting these diseases.
Discovering the mechanism by which prions become pathogenic is a crucial step in one day tackling these diseases, as it allows us to search for new drugs. Professor Alfonso De Simone
The research concerned prion diseases – a group of brain diseases caused by proteins called prions that malfunction and ‘misfold’, turning into a form that can accumulate and kill brain cells. These diseases can take decades to manifest, but are then aggressive and fatal.
They include Kuru, mad cow disease and its human equivalent Creutzfeldt-Jakob disease (CJD), and a heritable condition called fatal familial insomnia.
While the normal, healthy version of prions and the pathogenic (disease-causing) version have been characterised, the intermediate step, when one transforms to the other, was previously unknown.
Now, in a paper published today in Proceedings of the National Academy of Sciences, the research team have isolated this intermediate step, determining the mechanism that turns normal prions into their pathogenic form. The research was supported by Alzheimer’s Research UK.
Aggressive and devastating
Lead researcher Professor Alfonso De Simone, from the Department of Life Sciences at Imperial, said: “Prion diseases are aggressive and devastating, and currently there is no cure.
“Discovering the mechanism by which prions become pathogenic is a crucial step in one day tackling these diseases, as it allows us to search for new drugs. Now we know what we’re targeting, we know what features drugs need to have to stop prions becoming pathogenic.”
To investigate the misfolding of prions, the team worked with a mutant form of the prion protein that is found in people with inherited prion diseases. The mutant form is more aggressive, causing prions to transition faster to their pathogenic form. This allows the researchers to watch what happens more easily.
However, prions are difficult to isolate and purify from other proteins in sufficient quantities to study in detail. Lead author of the paper Dr Máximo Sanz-Hernández began investigating the problem as an undergraduate at Imperial, continuing until successful in his PostDoc with Professor De Simone.
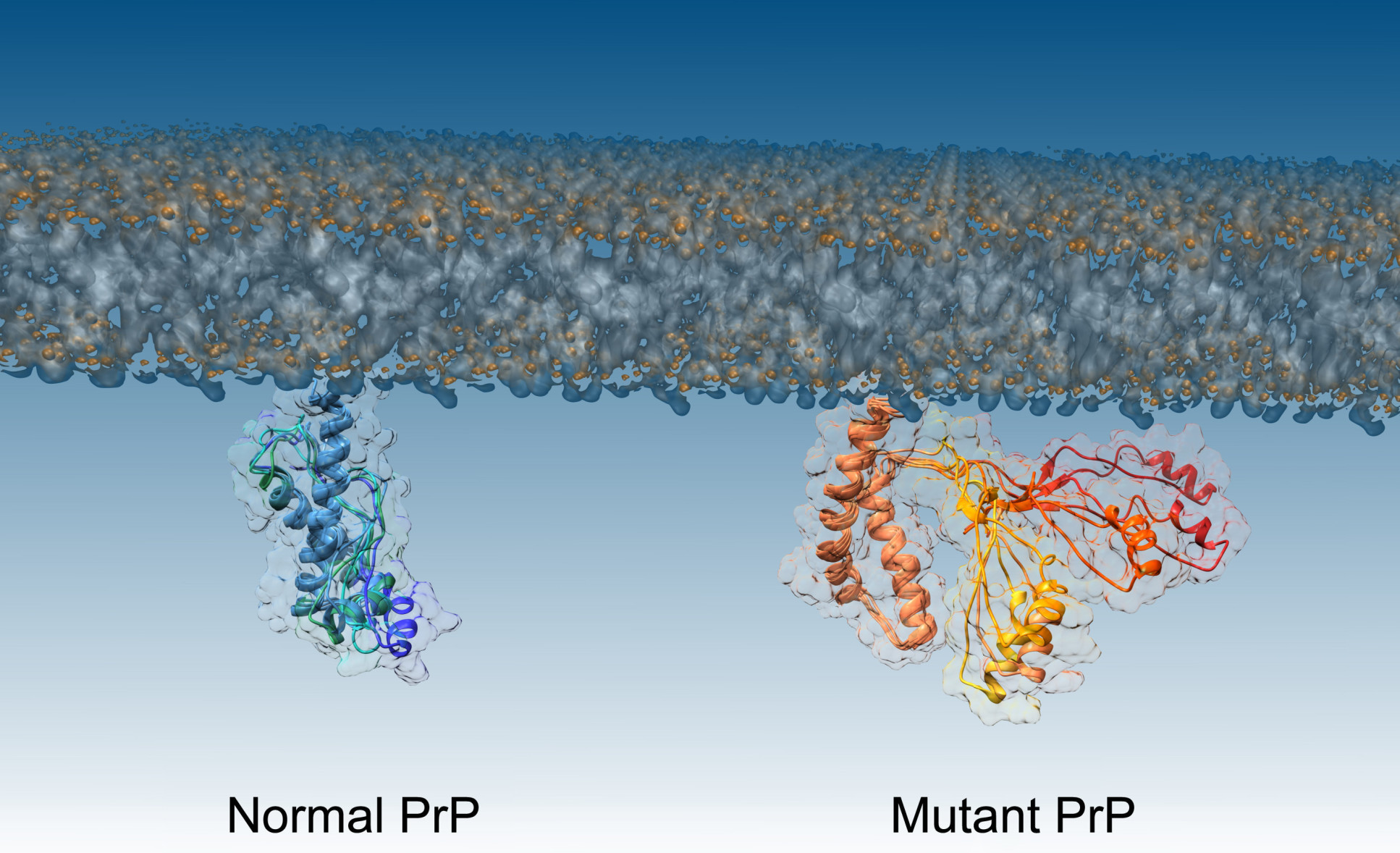
The team then used a technique called nuclear magnetic resonance spectroscopy combined with computational analysis to determine the structure of the intermediate step, identifying the molecular mechanism at work when the prion misfolds.
With this information, they also worked with the team at the University of Zurich who were able to produce antibodies that could target the mechanism. In a proof-of-concept study in the test tube, they were successfully able to block prions transitioning from the normal to the pathogenic form.
While in their current form, these antibodies would be too large to pass into the brain, the study shows it is possible to disrupt the mechanism, allowing researchers to move forward with designing new drugs.
Dr Sanz-Hernández said: “The intermediate stage of prion pathogenesis is so transient it’s like a ghost – almost impossible to image. But now we have a picture of what we’re dealing with, we can design more specific interventions that can one day potentially control these devastating diseases.”
Searching for drug compounds
Dr Rosa Sancho, Head of Research at Alzheimer’s Research UK, said: “This is early-stage research examining the short protein fragments, which can be highly unstable, short lived, and notoriously difficult to study.
“As the UK’s leading dementia research charity, we are pleased to fund this sophisticated work using biophysical and computational approaches to better understand the role fragments like this play in the development of disease. To identify new ways to reduce or combat these protein fragments in human disease we need to see sustained investment in dementia research.”
The researchers hope the information will allow drug researchers and pharmaceutical companies to scan their libraries of drug compounds for formulations that might be able to block the mechanism.
Any drug compounds would need extensive lab testing first to make sure they would be effective, small enough to pass into the brain, and safe, but the team hope that now the target is known, the search can be accelerated.
-
‘Mechanism of misfolding of the human prion protein revealed by a pathological mutation’ by Máximo Sanz-Hernández, Joseph D. Barritt, Jens Sobe, Simone Hornemann, Adriano Aguzzi and Alfonso De Simone is published in Proceedings of the National Academy of Sciences.
Article supporters
Article text (excluding photos or graphics) © Imperial College London.
Photos and graphics subject to third party copyright used with permission or © Imperial College London.
Reporter
Hayley Dunning
Communications Division