'Titin' gene mutations will help identify patients at risk of heart failure
by Sam Wong
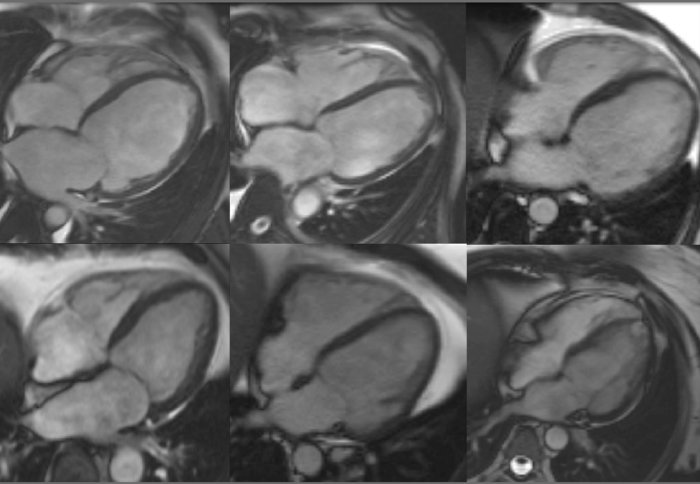
Cardiac magnetic resonance imaging (MRI) showing the hearts of six patients with dilated cardiomyopathy.
A new study has identified genetic mutations that cause the heart condition dilated cardiomyopathy (DCM), paving the way for more accurate diagnosis.
By sequencing the gene encoding the muscle protein titin in more than 5,000 people, scientists have worked out which variations are linked to disease, providing information that will help screen high-risk patients.
Titin gene mutations were previously associated with DCM, a leading cause of inherited heart failure, but many people have variations in the genetic code that are completely benign.
The new study, published in Science Translational Medicine, sorts the harmful from the harmless mutations, giving doctors a directory to interpret patients’ DNA sequences.
The information could also help researchers develop therapies to prevent or treat heart disease caused by titin mutations.
The study was led by researchers at Imperial College London and Royal Brompton & Harefield NHS Foundation Trust.

Above: Cardiac magnetic resonance imaging of the heart of a patient with dilated cardiomyopathy.
Around one in 250 people are estimated to have DCM. It causes the heart muscle to become thin and weak, often leading to heart failure.
Mutations in the titin gene that make the protein shorter, or truncated, are the most common cause of DCM, accounting for about a quarter of cases. But truncations in the gene are common – around one in 50 people have one – and most are not harmful, making it difficult to develop a useful genetic test.
The researchers sequenced the titin gene from 5,267 people, including healthy volunteers and patients with DCM, and analysed the levels of titin in samples of heart tissue. The results showed that mutations that cause DCM occur at the far end of the gene sequence. Mutations in healthy individuals tend to occur in parts of the gene that aren’t included in the final protein, allowing titin to remain functional.
Professor Stuart Cook, from the National Heart and Lung Institute and the Medical Research Council (MRC) Clinical Sciences Centre at Imperial College London, who led the study, said: “These results give us a detailed understanding of the molecular basis for dilated cardiomyopathy. We can use this information to screen patients’ relatives to identify those at risk of developing the disease, and help them to manage their condition early.”
The research was funded by the MRC, the British Heart Foundation, the Fondation Leducq, the Wellcome Trust, the National Institute for Health Research (NIHR) Royal Brompton Cardiovascular Biomedical Research Unit and the NIHR Imperial Biomedical Research Centre.

Secretary of State for Health Jeremy Hunt visiting the NIHR Royal Brompton Cardiovascular Biomedical Research Unit.
Professor Dudley Pennell, director of the NIHR Royal Brompton Cardiovascular Biomedical Research Unit, said: “This research reveals which genetic mutations are bad and which are there purely as bystanders. It will benefit patients with cardiomyopathy and enable us to reassure relatives who do not have the disease, allowing them to be discharged from clinic and preventing needless anxiety and unnecessary expensive tests.”
Professor Jeremy Pearson, Associate Medical Director at the British Heart Foundation, said: “Determining which mutations in titin are harmful and which are not has been difficult, in part because titin is one of the largest human proteins.
“This study defines, for the first time, a comprehensive list of mutations in the titin gene, which of these are associated with dilated cardiomyopathy, and which are harmless. This information will be extremely valuable for correct future diagnosis and treatment as we enter an era when many people’s genes will be sequenced.”
Reference: A.M. Roberts et al. ‘Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease.’ Sci. Transl. Med. 7, 270ra6 (2015).
Article supporters
Article text (excluding photos or graphics) © Imperial College London.
Photos and graphics subject to third party copyright used with permission or © Imperial College London.
Reporter
Sam Wong
School of Professional Development