Books

N-HETEROCYCLIC CARBENES: From Laboratory Curiosities to Efficient Synthetic Tools
Second Edition
Silvia Díez-González, Editor
In less than 20 years N-heterocyclic carbenes (NHCs) have become well-established ancillary ligands for the preparation of transition metal-based catalysts. This is mainly due to the fact that NHCs tend to bind strongly to metal centres, avoiding the need of excess ligand in catalytic reactions. Also, NHC‒metal complexes are often insensitive to air and moisture, and have proven remarkably resistant to oxidation. This book showcases the wide variety of applications of NHCs in different chemistry fields beyond being simple phosphine mimics. This second edition has been updated throughout, and now includes a new chapter on NHC‒main group element complexes. It covers the synthesis of NHC ligands and their corresponding metal complexes, as well as their bonding and stereoelectronic properties and applications in catalysis. This is complemented by related topics such as organocatalysis and biologically active complexes. Written for organic and inorganic chemists, this book is ideal for postgraduates, researchers and industrialists.
For the first edition of this volume, see: RSC Catalysis Series, 2010
Recent publications
63. [CuI(PPh3)3]-Mediated Arylation of 1,2,3-Triazoles
S. Lal, J. Qian, R. Pacheco-Muino, V. Kleene, M. J. Foulkes and S. Díez-González
ChemCatChem 2025, 17, e202500111
Part of the Celebratory Collection Women of Catalysis 2024

The arylation of 1,4-disubstituted triazoles with [CuI(PPh3)3] is reported. The reactions were carried out in relatively concentrated solutions of hot DMF in the presence of air and moisture. These conditions were suitable for a range of substrates, specifically triazoles containing a pyridyl group, which are of interest in diverse applications such as ancillary ligands or bioactive compounds.
62. Purifying the Air with Photocatalysis: Developing Bismuth Oxybromide/ Copper Phthalocyanine Composite Photocatalyst Filters with Enhanced Activity for NOxRemoval
O. Olaifa, P. Alimard, I. Itskou, F. Eisner, C. Petit, S. Díez-González and A. Kafizas
ChemPhotoChem 2025, 9 e202400346
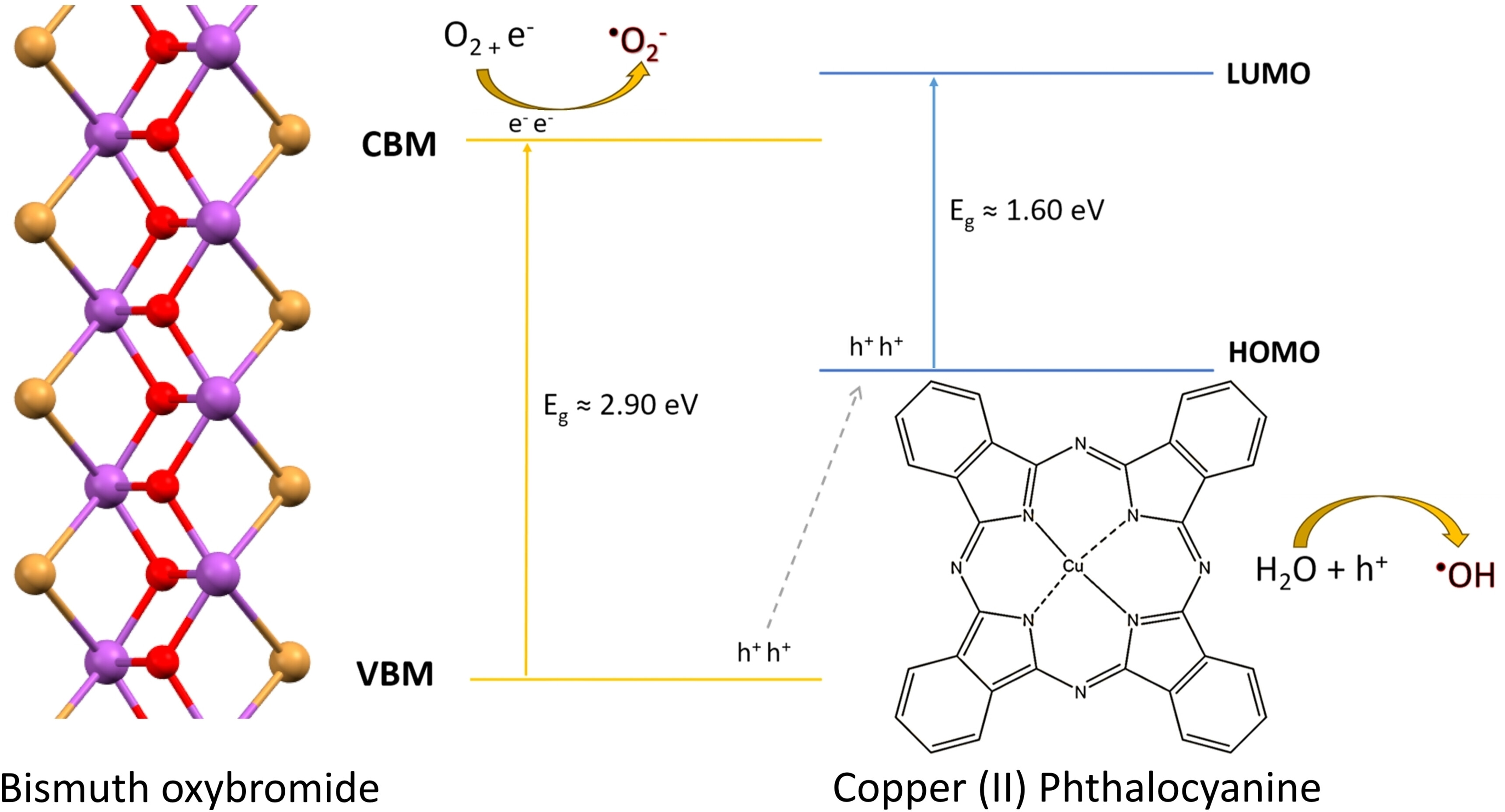
The utilization of photocatalysis is a promising new strategy for reducing the substantially high levels of nitrogen oxides (NOx) pollution in cities. In this work, we examine bismuth oxybromide (BiOBr) as a viable substitute due to its narrower band gap and high stability. Powders were synthesised using co-precipitation, solvothermal and hydrothermal synthesis methods, resulting in particles with various morphologies including microcubes, microspheres, microflowers, clusters and microsquares. Their photocatalytic activities being evaluated in accordance with ISO 22197–1 : 2016 protocol under UV and visible light. The samples exhibiting the highest performance were produced by co-precipitation, showing ~7 % NO and ~2 % NOx removal under visible light and ~19 % NO and ~10 % NOxremoval under UV light. The activity was further enhanced, by incorporating copper(II) phthalocyanine (CuPc) through an impregnation method, where the optimal loading of 0.01 mol% surpassed the activity of the benchmark photocatalyst TiO2 P25, with ~22 % NO and ~9 % NOx removal under visible light and ~40 % NO and ~23 % NOx under UV light. We anticipate that these BiOBr/CuPc photocatalyst filters can be applied within air purification systems and powered using less energy intensive visible light sources to remedy air pollution.
61. Bidentate NHC-Containing Ligands for Copper Catalysed Synthesis of Functionalised Diaryl Ethers
E. M. Richards, L. Casarrubios, J.M. D’Oyley, H. S. Rzepa, A. J. P. White, K. Goldberg, F. W. Goldberg, J. A. Bull and S. Díez-González
Adv. Synth. Catal. 2025, 367, e202400909
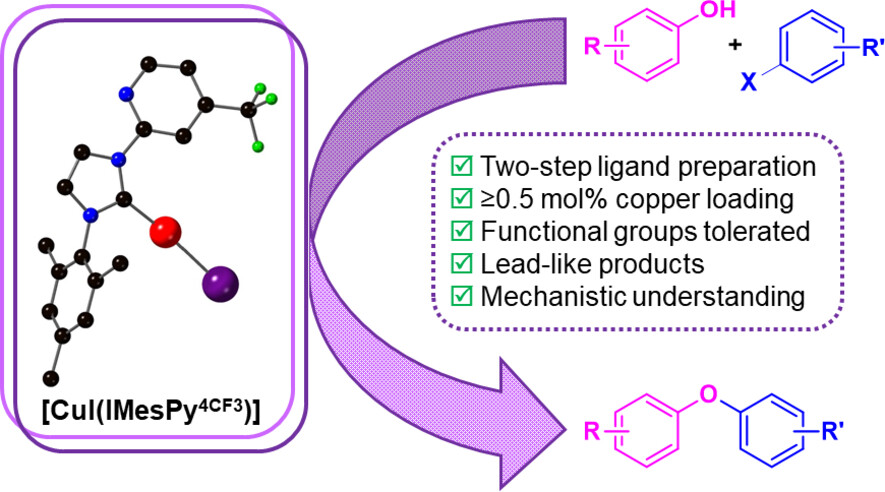
Diaryl and heteroaryl ethers are important and common motifs in drug discovery and agrochemicals. Here we present a catalytic system for the copper catalysed arylation of phenols with aryl halides. Bidentate N-heterocyclic carbene ligands provide a well-defined catalytic system that enable low loading of copper in effective coupling reactions (0.5 mol% Cu or lower). The reaction scope of aryl iodides and aryl bromides is developed, that is tolerant of functional groups and a library of biaryl ethers with lead-like properties is prepared. The presence of excess ligand (in a 1:3 ratio) is shown to be optimal to prevent catalyst degradation, presumably by avoiding decomplexation of the ligand. This is also supported by the catalytic results obtained with pre-formed copper complexes. The kinetics of the reaction are examined and shown to be first order in copper, supportive of a well-defined catalyst species, first order in phenol and aryl iodide, and zero order in base under the developed conditions. Computational studies performed using Minnesota MN15-L parametrisation method support an oxidative addition–reductive elimination pathway. Reductive elimination would be turnover limiting and occurr through an intimate ion pair intermediate.
Top Viewed Article 2025!

60. Cyclisation of Alkynyl Alcohols and Carboxylic Acids Mediated by N-Heterocyclic Carbene Copper(I) Complexes
C. Pimpasri, X. Luo, S. Yang and S. Díez-González
Adv. Synth. Catal. 2024, 366, 806–812
[Invited contribution to a special issue dedicated to Miquel A. Pericàs]
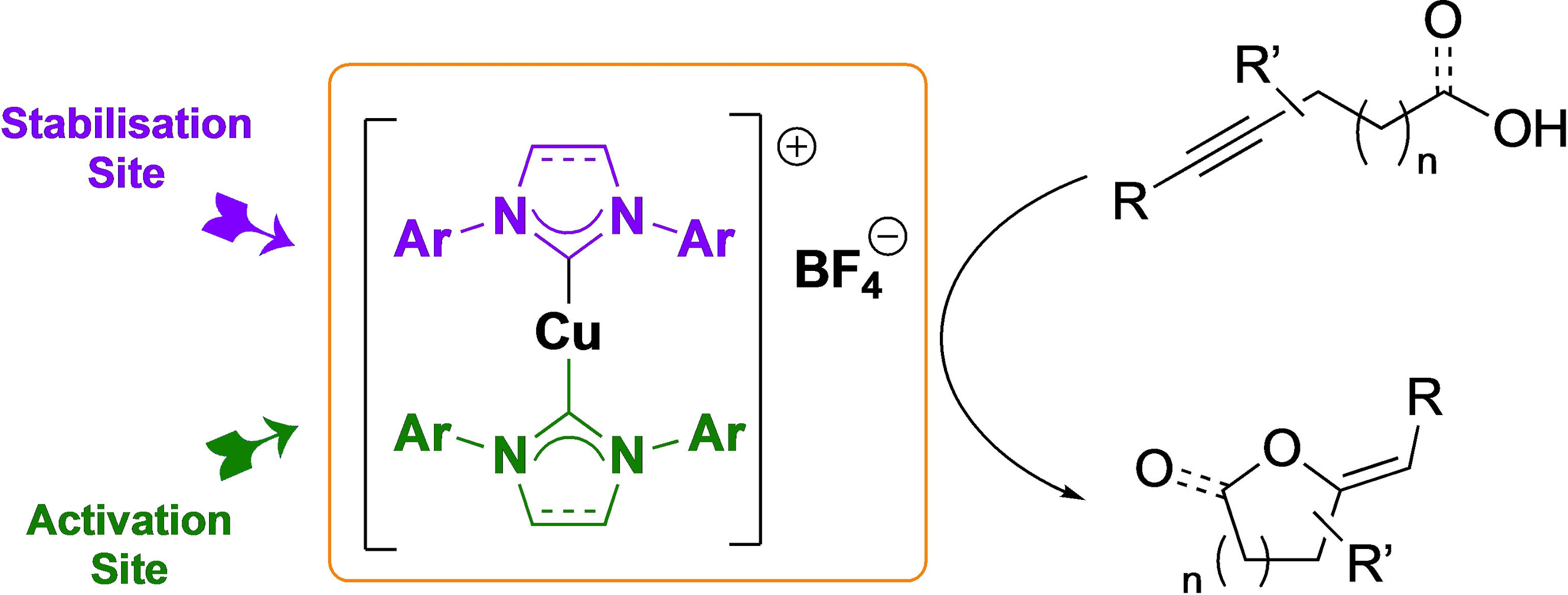
The cyclisation of alkynyl alcohols and carboxylic acids in the presence of a [Cu(NHC)2]X complex is reported. The reactions were carried out in technical grade toluene in the presence of air with a catalyst that is readily accessible. N-Heterocyclic carbenes are recognised for stabilising metal centres in a range of oxidation states, and this work further supports that NHCs can also be used as activation sites in air and moisture stable yet active catalysts.
59. Convenient Partial Reduction of CO2 to a Useful C1 Building Block: Efficient Access to 13C-Labelled N-Heterocyclic Carbenes
Phillips, N. A.; Sapsford, J. S.; Csókás, Kótai, D. B.; Perez-Tabarnero, I.; Díez-González, S.; Scott, D. J.; Pápai, I.; Ashley, A. E.
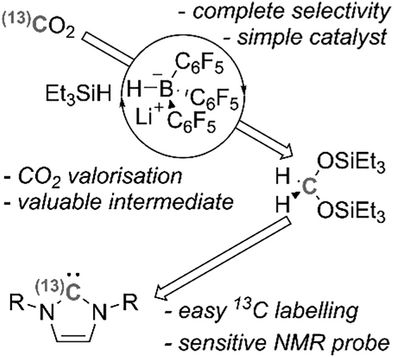 The selective, transition metal-free hydrosilylation of CO2 to CH2(OSiEt3)2 has been achieved under mild conditions and in high isolated yields (up to 90%) by using Et3SiH and the simple, easily prepared borohydride catalyst Li+[HB(C6F5)3]-. The resulting CO2-derived bis(silyl)acetal product -whose mechanism of formation has been interrogated through detailed computational and experimental studies- can be rapidly valorized through the facile synthesis of N-heterocyclic carbenes, via their corresponding imidazolium salts. By using relatively inexensive, isotopically enriched 13CO2 this protocol can be exploited to preare NHC isotopologues that are selectively 13C labelled at the key, ligation C2 position. This provides an electronically responsive 13C NMR spectroscopic handle with dramatically enhanced sensitivity, which can directly benefit reactivity studies in both organo- and organometallic catalysis, where NHC use is ubiquitous.
The selective, transition metal-free hydrosilylation of CO2 to CH2(OSiEt3)2 has been achieved under mild conditions and in high isolated yields (up to 90%) by using Et3SiH and the simple, easily prepared borohydride catalyst Li+[HB(C6F5)3]-. The resulting CO2-derived bis(silyl)acetal product -whose mechanism of formation has been interrogated through detailed computational and experimental studies- can be rapidly valorized through the facile synthesis of N-heterocyclic carbenes, via their corresponding imidazolium salts. By using relatively inexensive, isotopically enriched 13CO2 this protocol can be exploited to preare NHC isotopologues that are selectively 13C labelled at the key, ligation C2 position. This provides an electronically responsive 13C NMR spectroscopic handle with dramatically enhanced sensitivity, which can directly benefit reactivity studies in both organo- and organometallic catalysis, where NHC use is ubiquitous.
58. Straightforward and Efficient Deuteration of Terminal Alkynes with Copper Catalysis
Tarrach, X.; Yang, J.; Soleiman-Beigi, M.; Díez-González, S.

The mild and effective preparation of deuterated organic molecules is an active area of research due to their important applications. Herein, we report an air-stable and easy to access copper(I) complex as catalyst for the deuteration of mono-substituted alkynes. Reactions were carried out in technical solvents and in the presence of air, to obtain excellent deuterium incorporation in a range of functionalized alkynes.
57. Meyer-Schuster Rearrangement of Propargylic Alcohols Mediated by Phosphorus-Containing Brønsted Acid Catalysts
Radtanajiravong, L.; Peters, J.; Hummell, J.; Díez-González, S.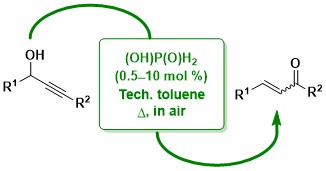
Org. Biomol. Chem. 2022, 20, 7338–7342
Commercially available (aqueous) hypophosphorus acid is an efficient catalyst for the synthesis of a,b-unsaturated carbonyl compounds from their corresponding propargylic alcohols. Reactions were carried out in technical toluene in the presence of air and in several instances the desired products were isolated analytically pure after a simple work-up.
56. Expedient Metal-Free Preparation of Aryl Aziridines via Thermal Cycloaddition Reactions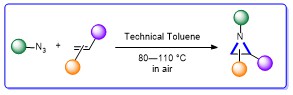
Sebest, F.; Radtanajiravong, L.; Kaukver, S.; White, A. J. P.; Díez-González, S.
Chem. Commun. 2022, 58, 3681–3184
A straightforward synthesis of aryl aziridines is reported from readily available azides and alkenes and using technical solvents in the presence of air. This methodology does not require any additives and the obtained compounds can be employed in ring opening and ring expansion reactions.
55. Cycloaddition Reactions of Azides and Electron-Deficient Alkenes in Deep Eutectic Solvents: Pyrazolines, Aziridines and other Surprises
Sebest, F.; Lachhani, K.; Pimpasri, C.; Casarrubios, L.; White, A. J. P.; Rzepa, H. S.; Díez-González, S.
Adv. Synth. Catal. 2020, 362, 1877–1886
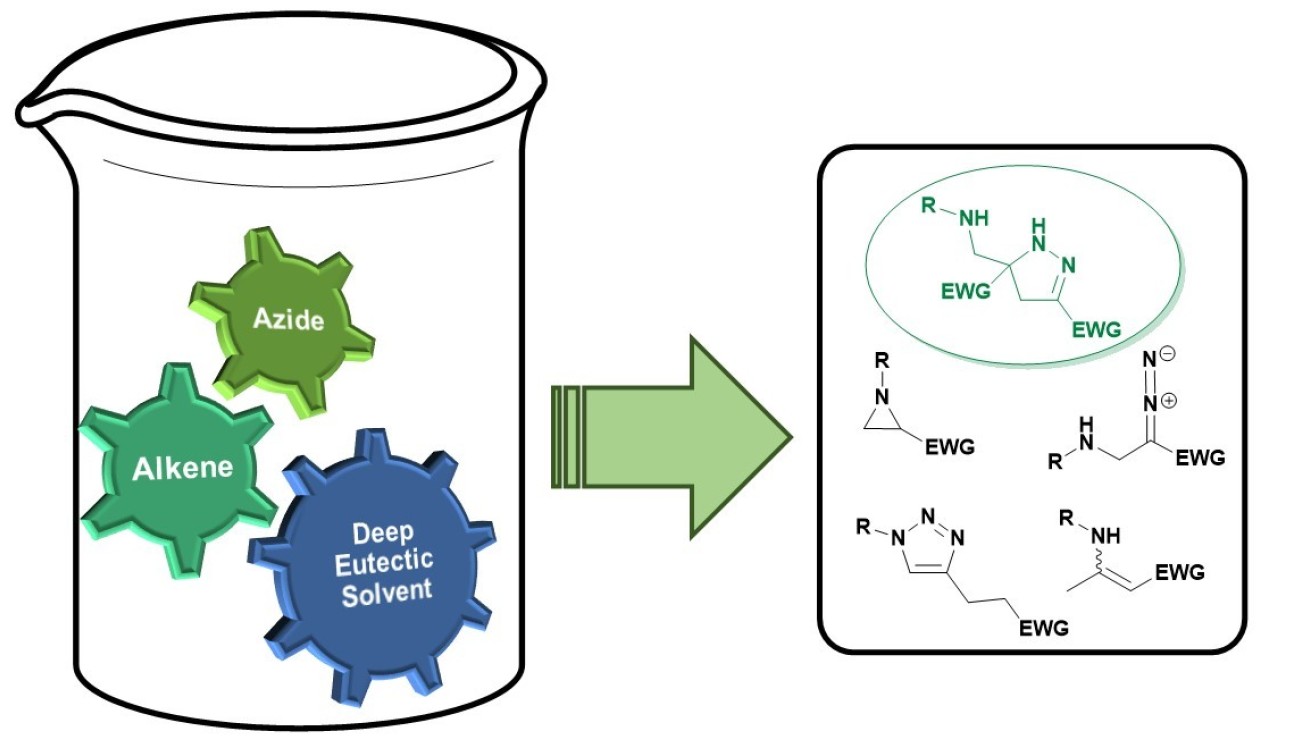
The reaction of organic azides and electron‐deficient alkenes was investigated in a deep eutectic solvents. A series of highly substituted 2‐pyrazolines was successfully isolated and their formation rationalised by DFT calculations. The critical effect of substitution was also explored; even relatively small changes in the cycloaddition partners led to completely different reaction outcomes and triazolines, triazoles or enaminones can be formed as major products depending on the alkene employed.
54. User-Friendly Copper-catalysed Reduction of Azides to Amines
Pimpasri, C.; White, A. J. P.; Díez-González, S.
Asian J. Org. Chem. 2020, 9,399-405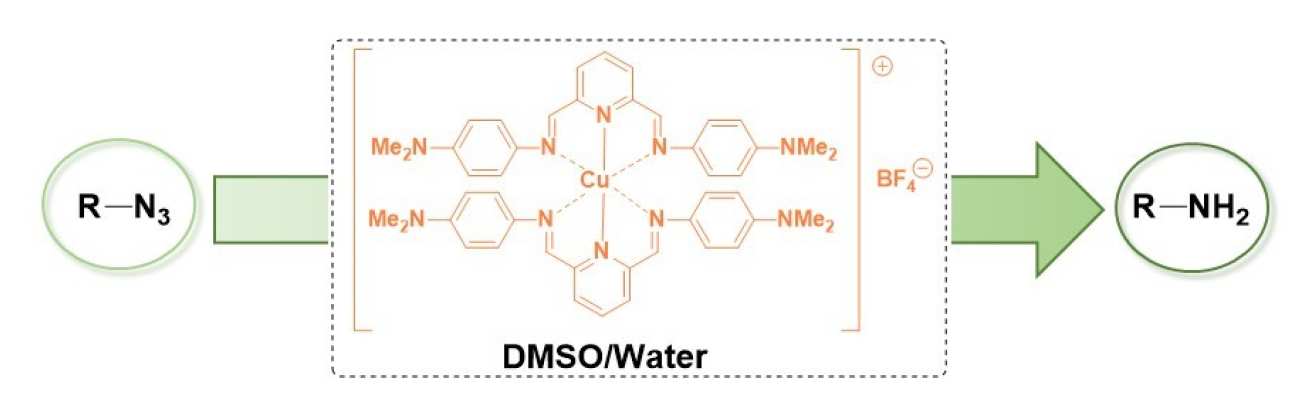
Three homoleptic copper(I) complexes have been prepared and applied to the reduction of organic azides. Under optimised conditions, complex [Cu(HPDIDMA)2]BF4 3 (HPDIDMA = 2,6-bis[N-(4-dimethylaminophenyl)carbaldimino]pyridine) could reduce a range of electron-poor azides in a mixture of DMSO and water without the need of an additional H-source.
53. Metal-Free 1,2,3-Triazole Synthesis in Deep Eutectic Solvents
Sebest, F.; Haselgrove, S.; White, A. J. P.; Díez-González, S.
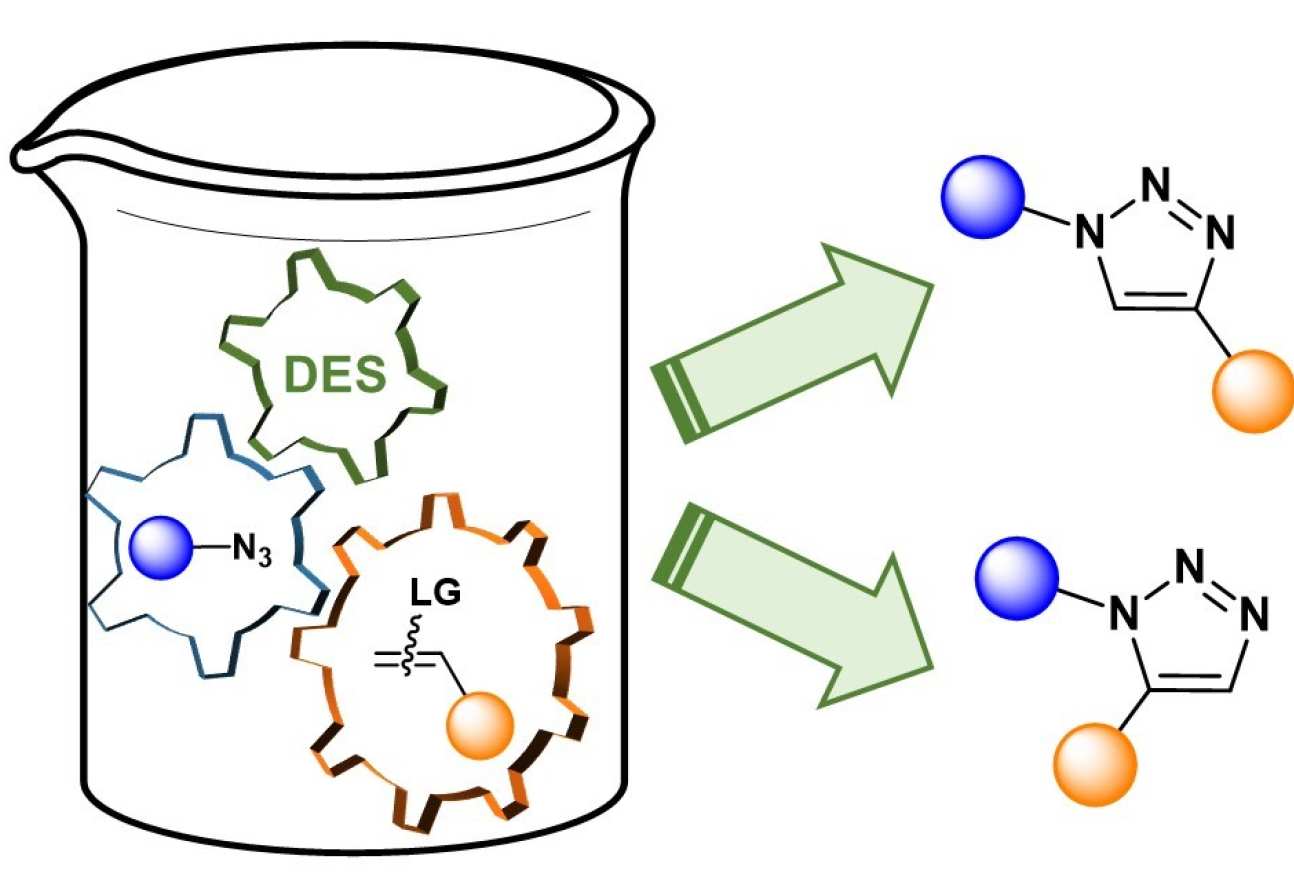
The metal-free regioselective preparation of 1,5- and 1,4-disubstituted triazoles is reported through a cycloaddition-elimination sequence. Reactions were carried out in environmentally friendly DES and pure products were isolated without the need of chromatographic techniques
52. Taming Brønsted Acid Reactivity: Nucleophilic Substitutions of Propargylic Alcohols with N-Nucleophiles Mediated by Phosphorus-Based Brønsted Acid Catalysts
Radtanajiravong, L.; Díez-González, S.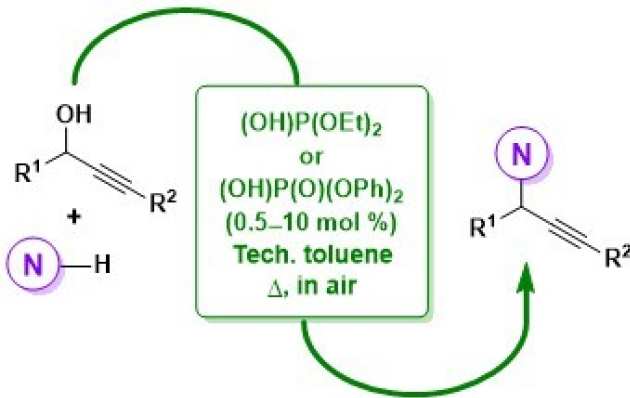
ACS Omega 2019, 4, 12300-12307.
The activity of diethyl phosphite and diphenyl phosphate in propargylation reactions with N-nucleophiles of varying basicity is presented. A careful choice of the reaction conditions minimized undesired rearrangements and arylation processes, typical side reactions with Brønsted acid catalysis. These systems are compatible with technical solvents and presence of air, and they are also applicable to C-, O-, and S-nucleophiles.
51. [Cu(NCMe)4](BF4): Commercially Available and User-Friendly Catalyst for Intramolecular Hydroamination Reactions
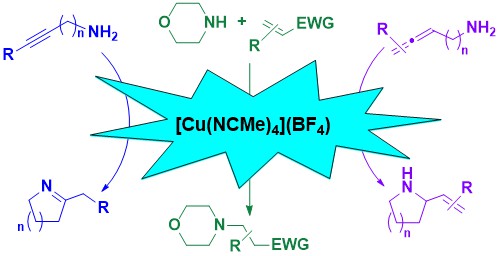
Zelenay, B.; Munton, P.; Tian, X.; Díez-González, S.
Eur. J. Org. Chem. 2019, 4, 4725–4730
The activity of a simple, commercially available copper salt, [Cu(NCMe)4](BF4) in intramolecular hydroamination reactions of alkynes and allenes is presented. Reactions were successfully carried out in technical acetonitrile in the presence of air. While attempts of alkene hydroamination failed, this catalysts was also found active in intermolecular aza-Michael reactions.
50. Cp*Fe(Me2PCH2CH2PMe2)(CHO): Hydride Shuttle Reactivity of a Thermally Stable Formyl Complex
Sapsford, J. S.; Gates, S. J.; Doyle, L. R.; Taylor, R. A.; Díez-González, S.; Ashley, A. E.
Inorg. Chim. Acta 2019, 488, 201–207
[Cp*Fe(Me2PCH2CH2PMe2)(CO)]+ [BArF24]− has been synthesised and characterised using single crystal X-ray diffraction, NMR and IR spectroscopies. Reduction of the CO ligand using Na[Et3BH] produces the corresponding neutral formyl complex Cp*Fe(Me2PCH2CH2PMe2)(CHO), that is very thermally stable, and which is attributed to the electron-releasing properties of the spectator ligands. This compound is a potent hydride donor which exists in equilibrium with [Et3BH]−, Et3B, and the structural isomer (η4-C5Me5H)Cp*Fe(Me2PCH2CH2PMe2)(CO), resulting from reversible hydride migration to the Cp* ligand.
49. Copper-Mediated Reduction of Azides under Seemingly Oxidising Conditions: Catalytic and Computational Studies
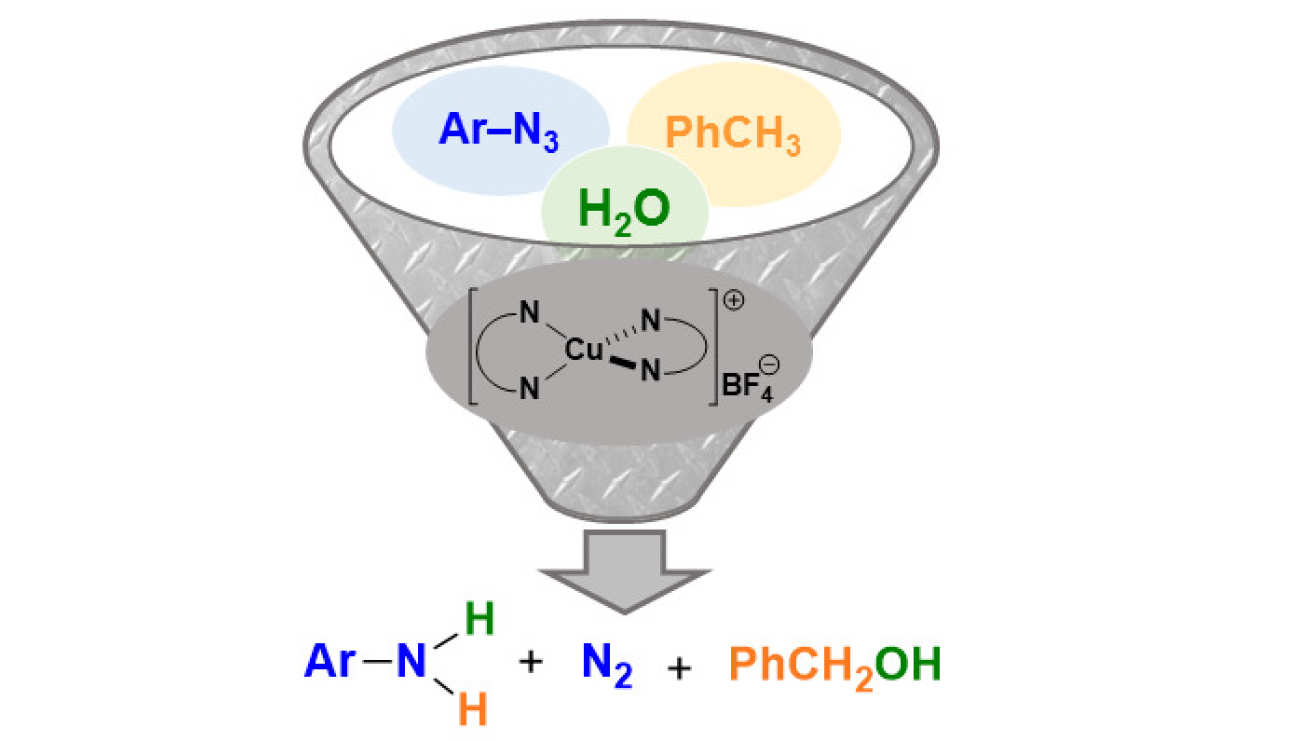
Zelenay, B.; Besora, M.; Monasterio, Z.; Ventura-Espinosa, D.; White, A. J.; Maseras, F.; Díez-González, S.
Catal. Sci Tech. 2018, 8, 5763–5773
The reduction of aryl azides in the absence of an obvious reducing agent is reported. Careful catalyst design led to the production of anilines in the presence of water and air. The reaction medium (toluene/water) is crucial for the success of the reaction, as DFT calculations support the formation of benzyl alcohol as the oxidation product. A singular catalytic cycle is presented for this transformation based on four key steps: nitrene formation through nitrogen extrusion, formal oxidative addition of water, C(sp3)–H activation of toluene and reductive elimination.
Contact us
Dr. Silvia Díez-González
Department of Chemistry
Imperial College London
London
W12 0BZ
Email: s.diez-gonzalez@imperial.ac.uk
Tel: +44 (0) 20759 49699