Quick way to create molecular cages could revamp search for new materials
A new way to predict the outcome of reactions could lead to faster discovery of new materials including for sensing, catalysis, and drug delivery.
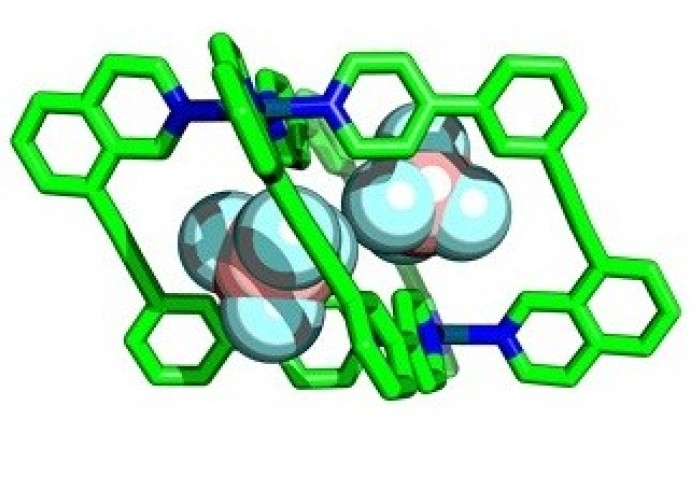
Enzymes perform essential chemical and biological processes by taking biomolecules into cavities within their structures and facilitating important reactions. Chemists have attempted to emulate this with ‘molecular cages’ – chemical structures that contain cavities that can bind smaller molecules within, called ‘guests’.
Now we can run some quick calculations, identify cages with properties that are useful, and be confident that we can synthesise them without any problems. Dr Jamie Lewis
These molecular cages have the potential to act as artificial enzyme-mimics and have been shown to accelerate important reactions such as the hydrolysis of amide bonds, degradation of toxins and a range of chemical transformations. Improving these reactions could one day drive the development of new technologies in areas such as chemical sensing.
However, it can be difficult for researchers to design structures that will be useful as well as successfully synthesised in the lab. Now, researchers from the Department of Chemistry at Imperial College London have used a computer-driven approach to predict the results of cage-building reactions with high precision.
This will help chemists select the ideal building blocks to prepare cages with desirable structures and properties before trying to synthesise them in the lab, minimising unsuccessful experimentation. The study is published today in Angewandte Chemie.
New building blocks
Currently, to simplify their synthesis, most cages and their cavities are highly symmetric. However, this limits the design of cages for potential guest molecules. This contrasts with natural enzymes’ ability to be highly selective in which molecules can bind with them.
Researchers at Imperial are developing ways to assemble cages with lower symmetry, allowing more guest-specific cavity shapes, by using more complex components in their construction. By using unsymmetrical building blocks, ‘wonky’ cages with interesting cavity shapes can be created.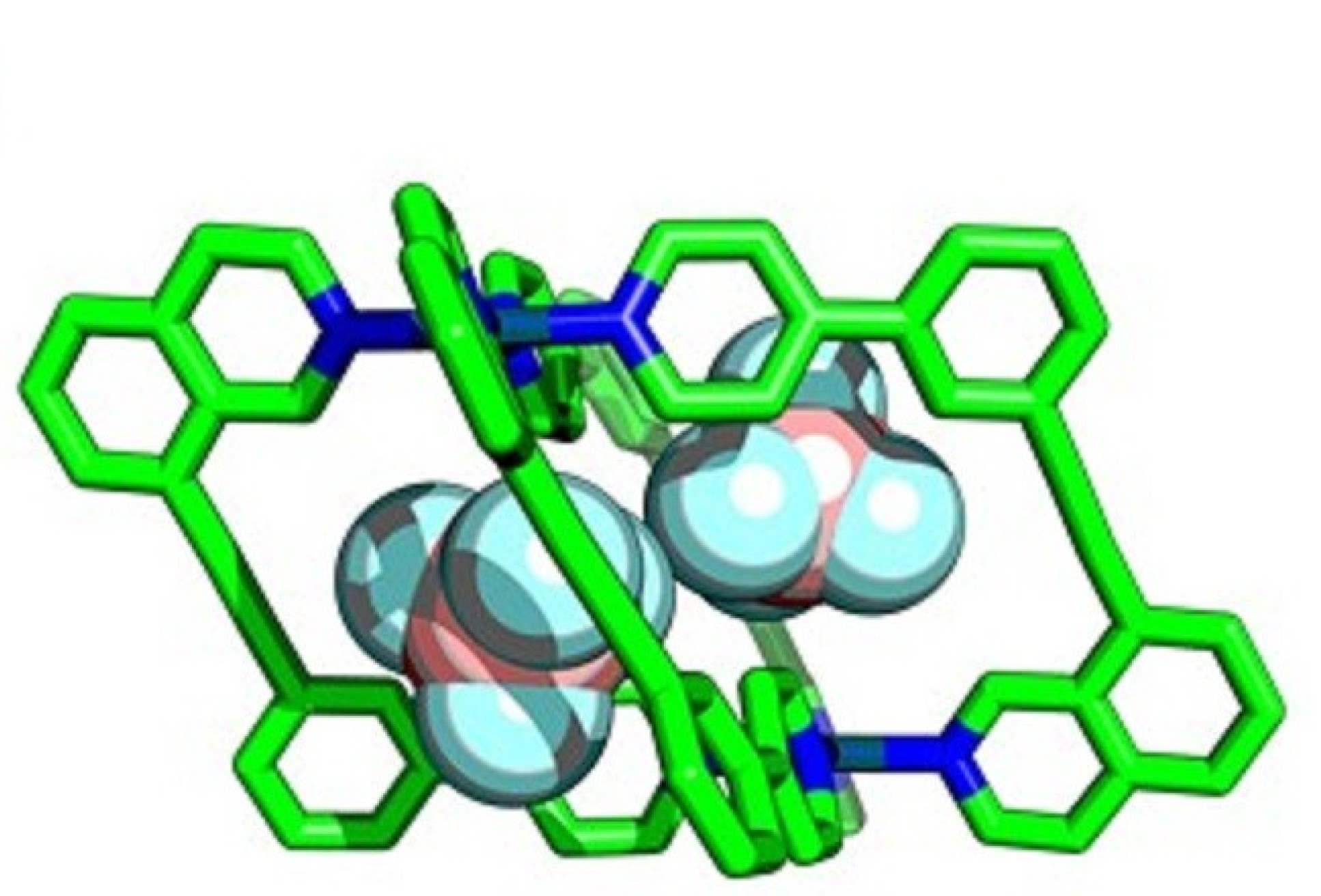
However, unsymmetrical building blocks are more difficult to create because the outcomes of the ‘self-assembly’ reactions needed to construct them are harder to predict. Failed reactions can result in the formation of an undesirable molecule, or even a mixture of products, rather than the single target structure.
All this means constructing new cages can be a time-consuming and costly trial-and-error process, with lots of wasted effort.
The new approach instead analyses computational models of potential cages to make predictions about their self-assembly. The predictions use the energy and geometry of the computationally constructed cages and are good guides for whether a self-assembly process will lead to a single structure. This information can then be used to select target cage molecules to prepare in the lab.
Study co-author Dr Jamie Lewis, from the Department of Chemistry at Imperial, said: “Previously, we’ve just had to get in the lab and try lots of things until something worked. Now we can run some quick calculations, identify cages with properties that are useful, and be confident that we can synthesise them without any problems.”
Prediction power
The team used software called stk, previously developed at Imperial, to build the computational models. As well as having large predicting power, the calculations are also very rapid, taking only a few hours on a common desktop PC.
Lead author of the study Dr Andrew Tarzia, from the Department of Chemistry at Imperial, said: “The efficiency of our approach is the key because it allows us to test on a computer many more building blocks in a week than could be tested in the laboratory and with more diversity too.”
Based on the computational data, the team selected a number of building blocks to synthesise in the lab. They found that the approach successfully predicted the experimental outcomes of the self-assembly process.
This allowed them to prepare several new low-symmetry ‘wonky cages’ that had never been synthesised before, and verified the usefulness of the calculations for predicting which molecules would form.
The team are now continuing to develop and improve this approach to computationally informed, efficient synthesis to access new molecular cages. With the ability to rapidly predict which cages can be readily prepared in the lab, they hope to use this to make novel materials with a wide range of applications in sensing, catalysis, gas storage and drug delivery.
-
‘High-throughput Computational Evaluation of Low Symmetry Pd2L4 Cages to Aid in System Design’ by Andrew Tarzia, James E. M. Lewis and Kim E. Jelfs is published in Angewandte Chemie.
Article supporters
Article text (excluding photos or graphics) © Imperial College London.
Photos and graphics subject to third party copyright used with permission or © Imperial College London.
Reporter
Hayley Dunning
Communications Division