Towards using quantum computing to speed up drug development
by Shang Yu
Researchers have developed a quantum processor that can perform crucial drug design methods, which could lead to more efficient drug development.
Drugs serve as a crucial means of treating diseases, and they mainly work by interacting with specific molecular targets within the body, thereby regulating or altering the functional status of these molecular targets. For example, a drug can bind with a viral protein and block the pathway of the virus entering human cells.
Quantum computing can simulate interactions between molecules more authentically, enabling us to predict the activity and safety of drug molecules more precisely during the drug design stage. Dr Raj Patel
In the current drug development process, the use of computational resources has become a key component. From molecular modelling and drug screening to pharmacodynamics prediction, large-scale computation and complex simulation are indispensable. These calculations often require a considerable amount of time and high-performance computing resources. So, can the recently popular quantum computing offer help with such drug design problems?
Researchers from Imperial College London, in collaboration with Zhejiang Lab and the University of Science and Technology of China, successfully developed a programmable Gaussian boson sampling light quantum processor, named “Abacus”, and adeptly applied it to two crucial drug design methods: molecular docking and RNA folding prediction. This accomplishment marks a significant step toward employing quantum computing to solve practical problems. The results are published in Nature Computational Science.
Dr Raj Patel, from the Department of Physics at Imperial, said: “With the rapid ongoing development of quantum computing technology, its application in drug development heralds a series of transformations and advancements. Quantum computing can simulate interactions between molecules more authentically, enabling us to predict the activity and safety of drug molecules more precisely during the drug design stage.
"This technology can also accelerate high-throughput screening of drugs, handle previously elusive complex biological systems, such as protein complexes, and promote cross-collaboration between physics, computational science, biology, and pharmacology.”
The quantum advantage
With the advancement of pharmaceutical science, more and more diseases and pathological conditions can be treated or alleviated through drug intervention. However, the discovery and development process of new drugs is both costly and time-consuming. For scientists to design effective and safe drugs efficiently, they need to have an in-depth understanding of the interactions between drug molecules and their target biological molecules.
Traditional computers have played a role in this regard, but simulating large, complex biomolecular systems, especially when involving quantum effects, dramatically increases the difficulty and complexity of computations. This is because the behavior of biomolecules is largely driven by quantum mechanical principles, and conventional computational methods often require approximations, which may lead to a loss of accuracy.
Quantum computers provide a new computational paradigm that allows us to simulate quantum systems directly and efficiently. This means it can capture the details of intermolecular interactions more authentically, providing more accurate data and insights for drug design. For complex molecular systems that conventional computing cannot efficiently handle, quantum computing might be the key.
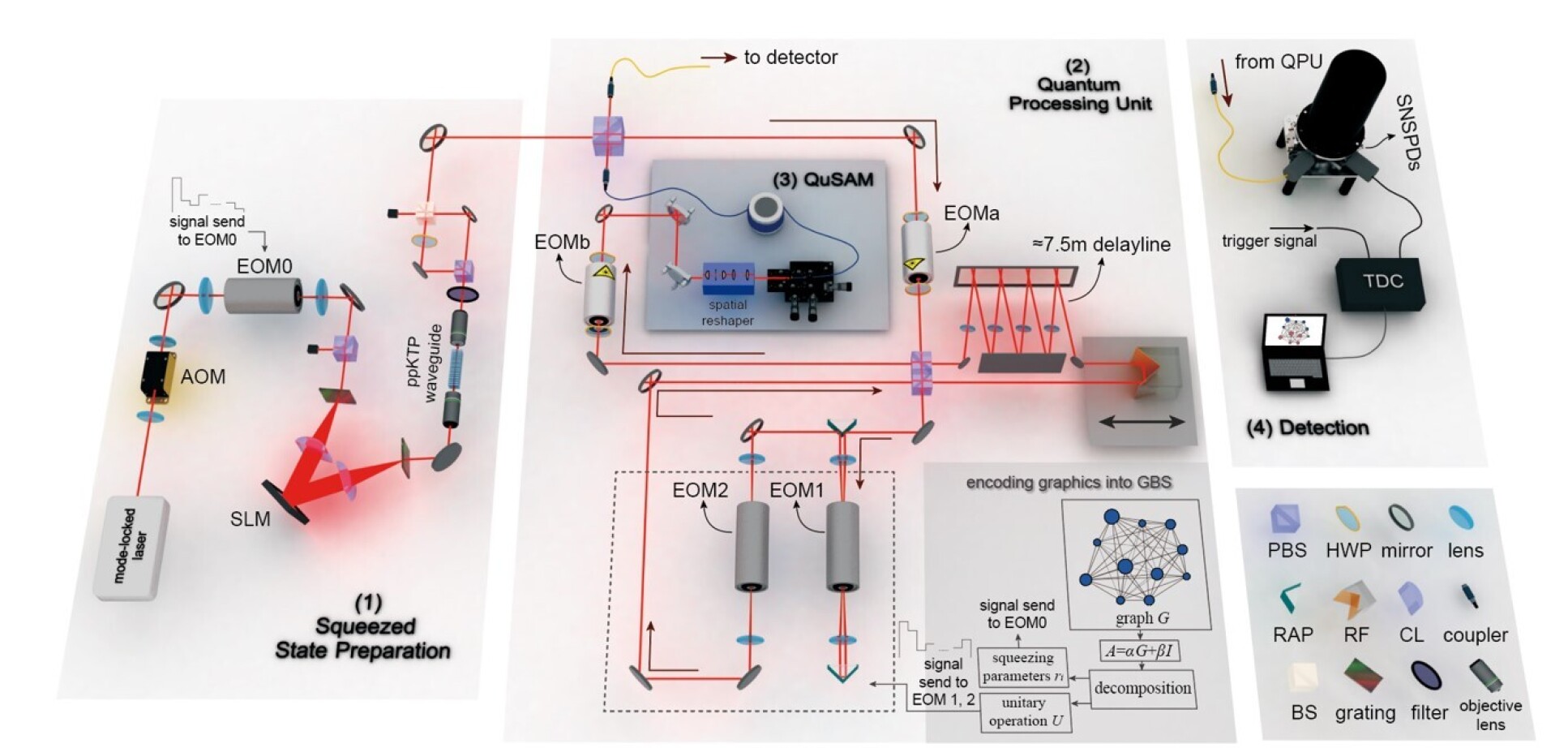
Dr Shang Yu, from the Department of Physics at Imperial, said: “Using quantum computers for drug design can help us find potential drug candidates more quickly, predict their biological activity more accurately, and optimize their efficacy and safety more effectively. Ultimately, this promises to accelerate the launch of new drugs, reduce R&D costs, and bring more and better treatment options to patients.
“Although based on current technology, quantum computing technology cannot be immediately applied to pharmaceutical companies for related production and research due to yet uncorrected loss and errors. However, in the long term, quantum computing has the potential to improve the efficiency of drug development, reduce overall costs, and drive the emergence of a series of new computing tools and algorithms. The integration of this technology is expected to lead drug development into a new era, bringing more efficient and innovative treatment plans to patients worldwide.”
Using Abacus
The team describe how they tested the Abacus system.
molecular docking
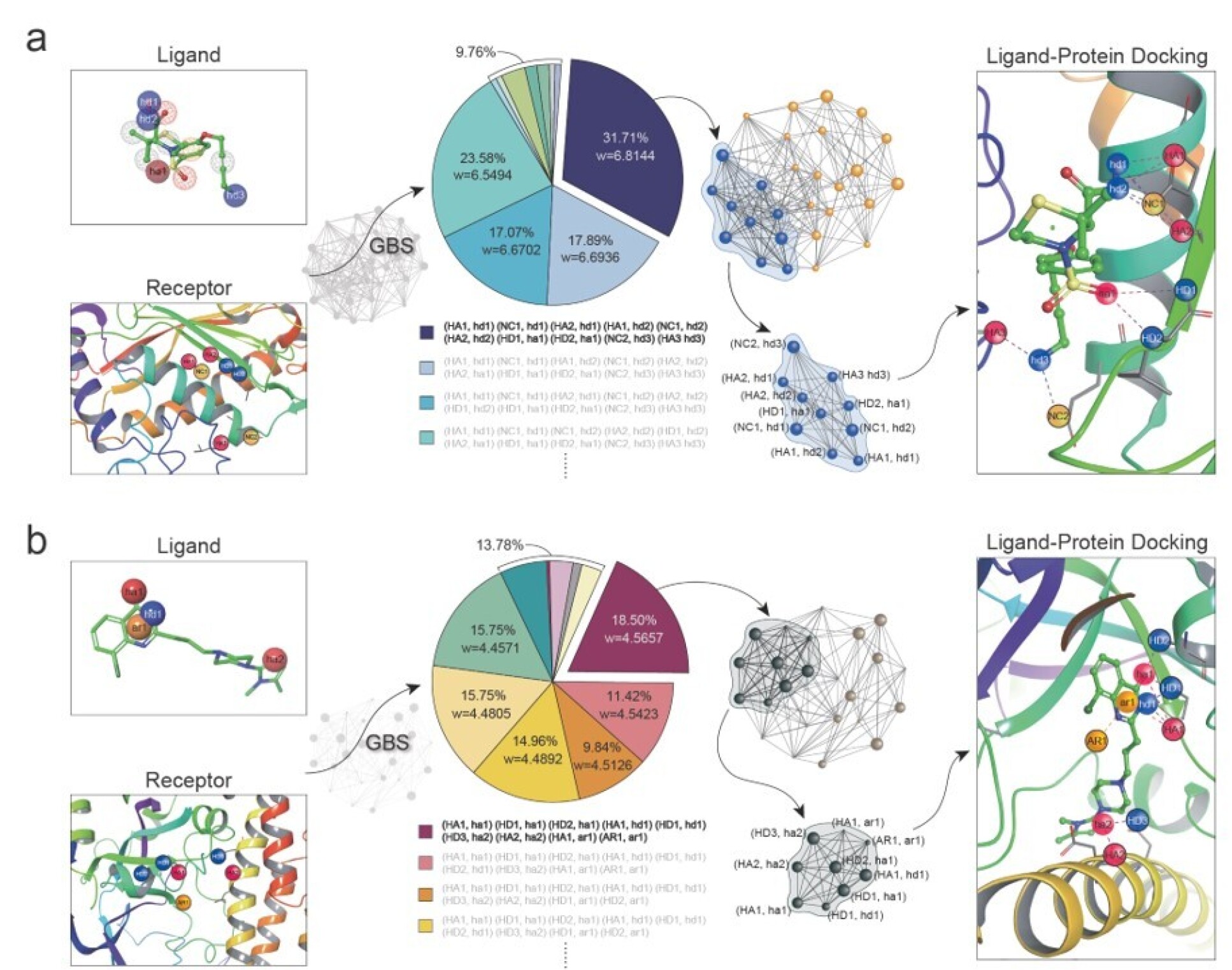
To validate that the “Abacus” can indeed be applied to real-world problems, we conducted tests on two commonly used drugs with known properties. Firstly, we modeled a compound of Poly (ADP-ribose) polymerase (PARP) and 8-chloroquinazolinone-based inhibitor (PARP-CQ) into a graph with 28 nodes. This represents a promising candidate drug for anti-cancer or certain Central Nervous System (CNS) diseases, such as Parkinson’s and Alzheimer’s.
Simultaneously, we constructed a 24-node interaction graph for α-tumor necrosis factor (TNF)-α convertase (TACE) and thiomorpholine sulphonamide hydrochloride inhibitor (TACE-TS), which are related to inflammatory diseases. We encoded the constructed graph structures into “Abacus” and utilized Gaussian Boson Sampling to perform the maximum fully connected subgraph search tasks on these graphs. Through the maximum fully connected subgraphs found, we can infer the optimal binding pose between the drug and viral protein.
RNA folding prediction
In drug design work, the molecular docking process is highly dependent on protein structure. In fact, many pathogenic proteins related to human diseases cannot be targeted by conventional small molecule drugs or large biomolecules. In recent years, nucleic acid drugs have garnered attention in the pharmaceutical field because they may become a potential solution to overcome the limitations of existing target drugs and treat previously “untargetable” diseases.
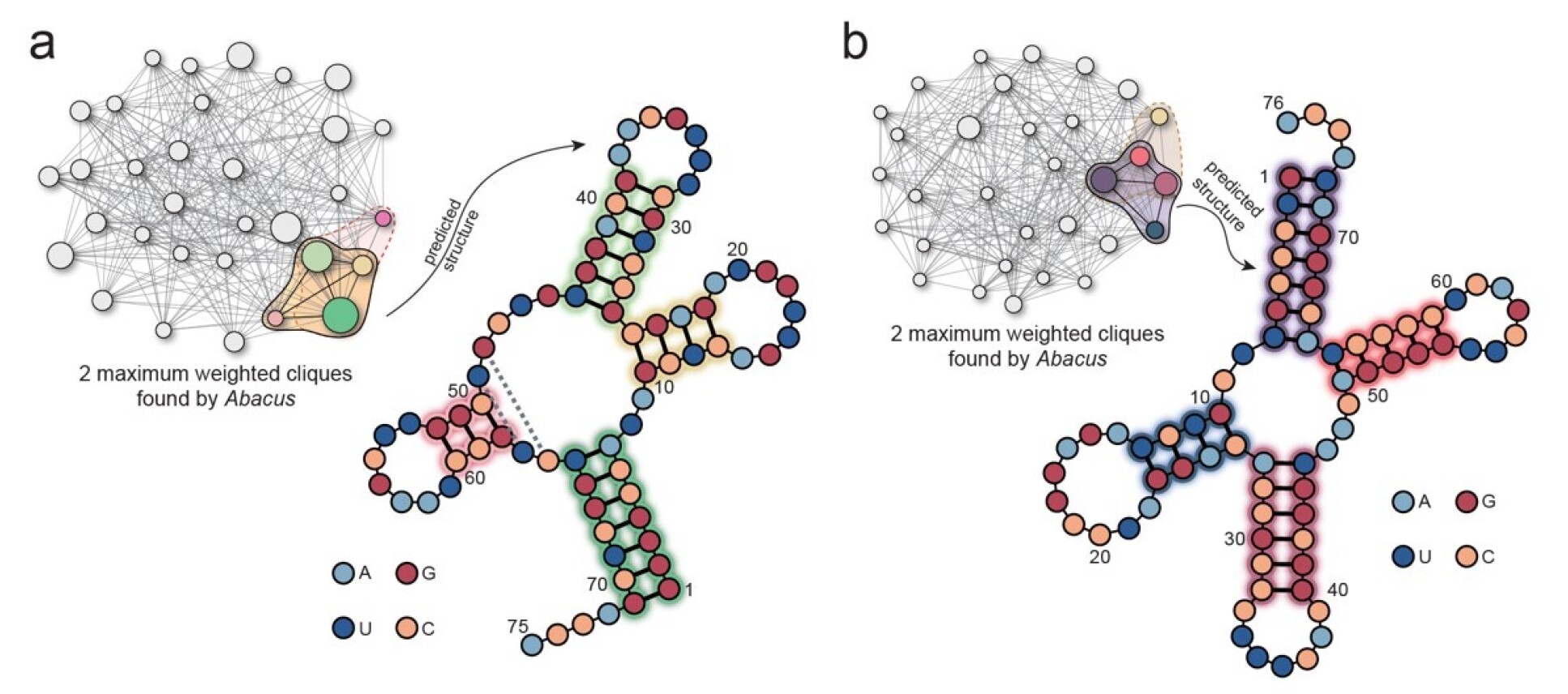
Predicting RNA structures has become an important task in discovering these nucleic acid drugs, as it can help identify potential drug targets and predict the interactions of small molecule drugs with RNA molecules. In exchanges with Dr Mengyi Tang and Professor Sung Ha Kang from the Georgia Institute of Technology, researchers found that the “Abacus” can also be used to predict the folding of RNA sequences.
This method involves modeling the RNA sequence into a weighted full stem graph, and then encoding it into the “Abacus”. Such a weighted full stem graph captures all possible folding information of the RNA sequence, where each node represents a possible stem in the sequence, and edges represent their coexistence relationship. The weight of each node corresponds to the length of the stem it represents. Then, the prediction of the optimal RNA folding structure can be obtained by finding the maximum fully connected subgraph in the weighted full stem graph. To demonstrate the efficacy of our programmable Gaussian Boson Sampler in solving this problem, we conducted experiments on two different RNA fragments on the “Abacus”. shown above.
-
“A Universal Programmable Gaussian Boson Sampler for Drug Discovery”, is published in Nature Computational Science.
Article text (excluding photos or graphics) © Imperial College London.
Photos and graphics subject to third party copyright used with permission or © Imperial College London.
Reporter
Shang Yu
Department of Physics