Project 3
- Development of single-ionization Algebraic Diagrammatic Construction theory to yield energy gradients needed for nuclear dynamics calculations.
- Extension of our Complete Active Space Self-Consistent Field code to the case of sudden single ionization.
- Measurement of the effects of charge migration using ~ 1fs resolution chromophore specific VUV pump + IR-field probe, and VUV pump + XUV probe in medium-sized organic molecules and in biomolecular building blocks.
- Measure the effects of charge migration using an Auger marker method to start localized site-specific dynamics.
Current Research
- Single-photon laser enabled Auger decay spectroscopy for measuring attosecond electron hole dynamics
- Ultrafast ICD in endohedral fullerenes mediated by dipole and multipole plasmon excitations
- The role of attosecond electronic coherence in molecular dissociation
- Angular distributions for correlation-assisted tunnelling
- Probing ultra fast molecular dynamics with X-ray free-electron lasers
- Partial covariance mapping technique for probing electron dynamics in molecules
- Coupled single-shot electron VMI and ion time of flight detector.
- Coherent Superposition of Electronic States and Electronic Dynamics in Molecules Studied using the Ehrenfest Method
- Ultrafast hole dynamics measured by few femtosecond x-ray pump-probe Auger spectroscopy
- Effect of nuclear dynamics on charge migration in glycine
- Heteroatomic Rare-Gas Clusters
Introduction
We consider glycine, the simplest amino acid, ionised from an inner-valence orbital using an XUV pulse in the sudden regime. This ionisation regime does not lead to a single eigenstate of the ion, but rather a superposition of eigenstates. These non-stationary states evolve in time often leading to electron hole dynamics on the sub-femtosecond to few-femtosecond time scales [1, 2]. Here we propose a new time-resolved attosecond technique, single-photon laser-enabled Auger decay (spLEAD) spectroscopy, that uses a VUV ionizing probe and is particularly suited to characterising hole migration dynamics in the inner valence energy region.
Theory
Recently, Kapteyn and Murnane [3] have investigated Laser-Enabled Auger Decay (LEAD) in the multi-photon regime. Here, we show that if considered in the single-photon regime, the LEAD process [shown in Fig. 1. for (2s−1)Ne+] provides valuable insight in electron correlation and the nature of inner- valence ionised states.
We also show that the spLEAD technique can map the dynamics that occur in inner-valence ionised molecules, such as hole migration [1, 2], onto a double ionisation probability as a function of the time delay between XUV pump-VUV probe. The temporal evolution, of the superposition of eigenstates after sudden ionisation, can be characterised by the survival probability S(t) of the initial state. Short VUV pulses can be used to map the survival probability onto the double ionisation yield P(t) as a function of VUV pulse delay relative to the XUV ionising pulse, as the doubly charged ion signal comes exclusively from the spLEAD of target inner-valence ionised states.
Ionising from the 11A' orbital in glycine results in oscillatory dynamics involving the 11A' and 12A' ionised 1h configurations [4]. Our ab initio calculations [5] demonstrate excellent correspondence be- tween the double ionization probability with the survival probability, shown in Fig. 2, indicating that this two-state hole migration process is exceptionally well resolved using the proposed spLEAD probe.
Current researchers
B. Cooper and V. Averbukh.
References and relevant links
[1] L. S. Cederbaum, J. Zobeley, Chem. Phys. Lett. 307, 205 (1999).
[2] J. Breidbach and L. S. Cederbaum, J. Chem. Phys. 118, 3983 (2003).
[3] P. Ranitovic et al. Phys. Rev. Lett. 106, 053002 (2011).
[4] A. I. Kuleff and L. S. Cederbaum, Chem. Phys. 338, 320 (2007).
[5] B. Cooper and V. Averbukh, Phys. Rev. Lett. 111 083004 (2013)
Introduction
Interatomic (Intermolecular) Coulombic Decay- ICD for short [1], is an mechanism for the decay of inner- valence vacancies that do not possess enough energy to decay via the Auger process [2]. ICD proceeds in clusters of atoms (or molecules) where the presence of neighbouring species lowers the double ionization threshold of the cluster by allowing the positive charge to spread over different cluster subunits.
An isolated ion with an inner-valence vacancy whose energy is lower than the double ionization threshold of the ion decays via photo-emission, which is a very slow process with a lifetime in the nanosecond regime, compared to lifetimes of 10’s to several 100’s of femtoseconds that ICD has to offer when this ion is part of a cluster. ICD involves recombination of an outer-valence electron on the ionized species into the inner-valence vacancy, accompanied by ionization of a valence electron on a neighbouring cluster component, mediated by the Coulomb potential between the two electrons. ICD becomes ultrafast in endohedral fullerenes due to the many available decay channels. Here we study ICD in [2s−1]Ne+ @C60 , a 2s-ionized neon inside the hollow structure of the football-shaped C60 fullerene.
Theory
The energy transferred to the fullerene by the recombining neon ion is in the region of the s o-called giant plasmon [3] − a collective excitation of active electrons in C60. However, the initial theoretical work [4] considers only the central (i.e. symmetric) geometry of this system. Effect of non-central neon position inside the fullerene must be considered in order to gain a full picture of the process. The displacement of the trapped neon ion from the centre of C60 lowers the symmetry of the system. This may allow ICD energy to excite multipole plasmon states in C 60, which in turn can affect the rate of ICD [5].
In order to investigate how efficiently ICD in the displaced system excites multipole plasmon states in C60, we use Wigner-Weisskopf theory and multipole expansion of the Coulombic repulsion. By expanding the displaced Neon orbitals in terms of an infinite sum of spherical harmonics, whose coordinates are measured with respect to the c entre of C60[6], we can obtain the strength of excitation of each C60 plasmon as a function of the displacement (Fig. 1).
This was followed by numerical calculation of ICD rate, Γ, and investigation of its behaviour when Ne gets displaced from the centre. Meanwhile we made use of the SCF (Self-Consistent Field Theory) and MP2 (based on second-order Møller-Plesset perturbation theory) modules of the quantum chemistry software MOLCAS [8] in order to simulate the energy of the system and its dependence on displacement of the central atom. Fig. 2 shows both these results on one graph.
Current researchers
L. Bahmanpour and V. Averbukh.
References and relevant links
[1] F. Krausz, M. Ivanov, Rev. Mod. Phys. 81, 163-234 (2009).
[2] B. Crasemann, ”Atomic Inner-shell Physics”, (1985).
[3] I. V. Hertel, et al. Phys. Rev. Lett. 68, 784-787 (1992).
[4] V. Averbukh and L. Cederbaum, Phys. Rev. Lett. 96, 053401 (2006).
[5] N. Ju, A. Bulgac, J. W. Keller, Phys. Rev. B. 48, 9071-9079 (1993).
[6] H. Silverstone, J. Chem. Phys. 47, 537 (1967).
[7] A. Bug, A. Wilson, G. A. Voth, J. Phys. Chem. 96, 7864-7869 (1992).
[8] F. Aquilante et al., Journal of Computational Chemistry, 31, 224, (2010).
[9] K. Kaufmann, W. Baumeister, M. Jungen, J. Phys. B. 22, 2223 (1989).
Introduction
In molecules electronic and nuclear degrees of freedom are always correlated, but due to different timescales of their evolution, effects of electronic coherence on nuclear motion are usually not observed. However, we show that electronic coherence can significantly influence nuclear dynamics when interference of paths associated with vibrational motion along different potential energy surfaces becomes important.
Theory
We consider attosecond XUV pump IR probe setup. A N2 molecule is first ionized by an XUV pump pulse. Ionization creates a coherent superposition of two excited electronic states in N2+ ion. The dissociation of the molecular ion then takes place over two different potential energy surfaces. Interaction with IR field ensures that both of them lead to the same dissociation limit. The fragment spectrum obtained depends significantly on how the two paths interfere.
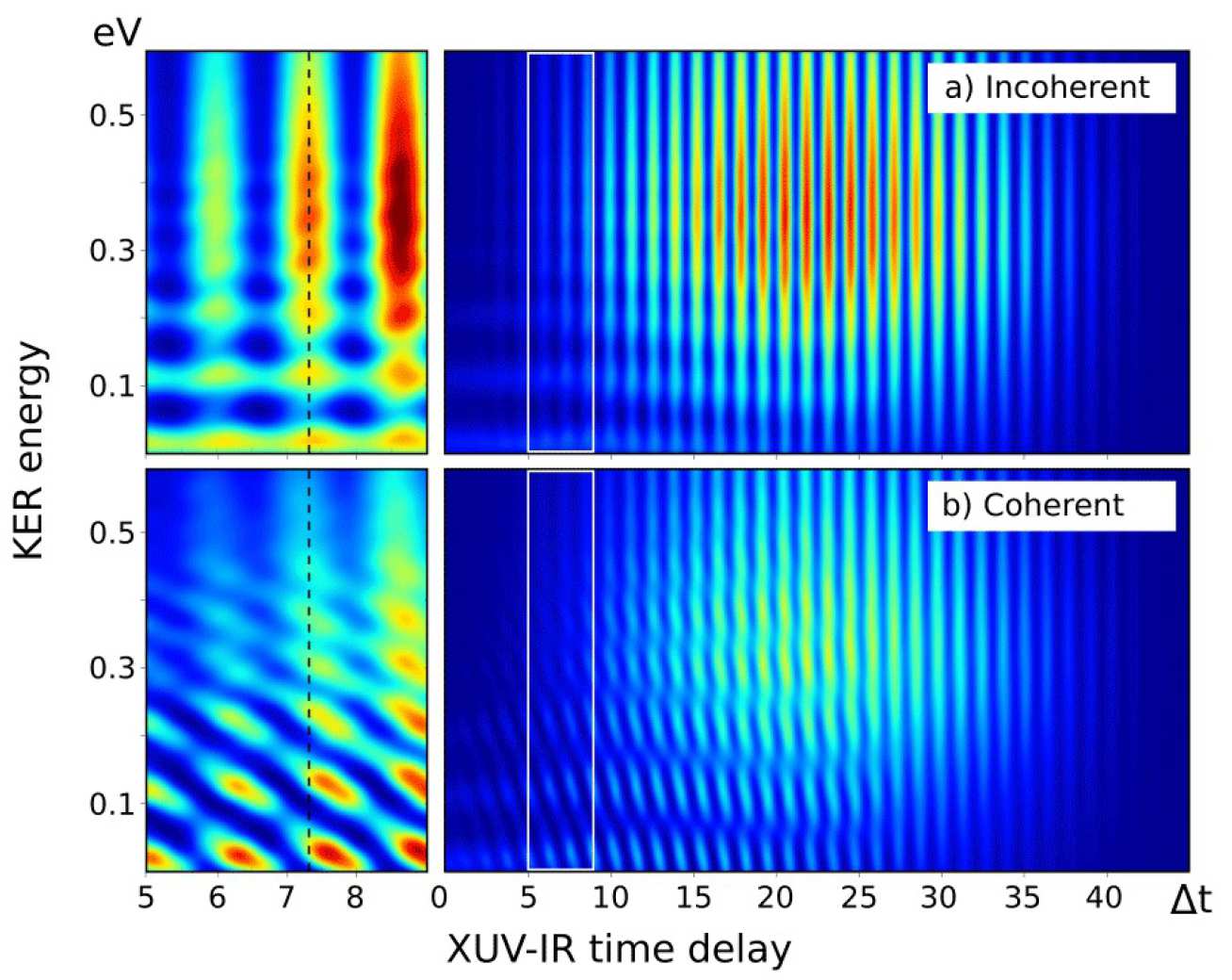
Interference is observed if the continuum electron wavepackets correlated with each of the paths have an overlap. This condition is set during ionization, which happens in a fraction of a femtosecond and is given by the length of the XUV pump pulse. However, it determines the appearance of molecular dissociation spectrum hundreds of femtoseconds later. Therefore the final spectrum observed is an interplay between attosecond electronic and femtosecond molecular dynamics.
When one of the pathways involves a direct and the other a sequential transition to a dissociative state, the phase acquired along the propagation on sequential path creates a chirp in the interference pattern and introduces additional phase in a RABBITT type me asurement on molecules. It can be identified in an XUV-IR time delay scan and can potentially encode information on the initial wavepacket and the potential energy surface.
Current researchers
L. Medisauskas, S.Patchkovskii1, and M.Ivanov.
1SIMS NRC Canada
Introduction
Electron-electron correlations and interactions are crucial concepts in the description of atoms and molecules which have only been included in studies of strong-field ionization since relatively recently. In the mean field, the interaction with the ionic core is an important perturbation on the laser-induced motion, slowing the electron down as it departs [1], but this neglects the correlations with the core. As an operator on the ionic state space, it has been treated perturbatively to describe excitations in the core [2, 3, 4] and multiple ionization [5].
A more formal treatment of the excitation of the core has been developed recently [6], indicating two distinct pathways leading to each excited ionic core state: tunnelling from a deeper orbital than HOMO and inducing transitions in the core, even when the electron is still inside the tunnelling barrier. The former suffers from an exponential penalty on a bigger barrier while the latter is suppressed by small interactions with a far-away core [7, 8], so the two can be comparable.
Theory
We analyse the angular distributions corresponding to these two mechanisms in detail, looking for qualitative signatures between them that will help single out the contribution from the correlation-assisted channel and reveal structural information of the initial and final states and of the transition itself.
Our results give a suitably accurate treatment of the evolution up to first order in the correlation interaction. The structure and symmetries of the original orbital and the transition have a strong influence on this signal, which can in fact differ structurally from the direct tunnelling one.
For example, perpendicular transitions of the type Π → Σ impose a central hole on the wavefunction as it departs, which translates into a ring-like structure reminiscent of Fraunhofer diffraction. The leaving electron has angular momentum Lz= h-bar, as does the ionic core; in the interaction the electron must ‘wind down’ the core and return its angular momentum through a reaction torque, which creates a central lobe. Lateral lobes appear through interference effects.
The central lobe is completely absent, on the other hand, from the semi-classical approximation to this spatial distribution (red in Fig. 1(c)), since that would require on-axis trajectories forbidden by the original orbital. We describe the necessary corrections to this approximation in the cases where it fails.
Thus we find that relatively complex, different wavefunctions can result from the two mechanisms even for simple initial orbitals. The final wavefunction is their coherent sum, which can accentuate or diminish the differences between them; this opens the hope of finding ways to control this mixture and thus shape the outgoing wavefunction.
Current researchers
E. Pisanty and M. Ivanov
References and relevant links
[1] O. Smirnova, M. Spanner and M. Ivanov. Phys. Rev. A 77, 033407 (2008).
[2] I. V. Litvinyu k et al. Phys. R ev. Lett. 94, 033003 (2005).
[3] B. A. Zon. J. Exp. Theor. Phys. 91, 899 (2000).
[4] J. P. Farrel et al. Phys. Rev. Lett. 107, 083007 (2011).
[5] A. S. Korn ev, E. B. Tulenko and B. A. Zon. Phys Rev. A 79, 063405 (2009).
[6] L. Torlina et al. Phys. Rev. A 86, 043409 (2012).
[7] L. Torlina and O. Smirnova Phys. Rev. A 86, 043408 (2012).
[8] Z. B. Walters and O. Smirnova. J. Phys. B 43, 161002 (2010).
Introduction
An entirely new perspective on ultrafast dynamics in small molecules has been provided by the advent of innovative light sources such as X-ray FELs. Among these the Linac Coherent Light Source (LCLS) is an ideal experimental tool as it is capable of producing few-femtoseconds short pulses in the soft X-ray range of the electromagnetic spectrum and therefore it gives access to processes which unfold on a femtosecond time scale. In the domain of femtochemistry the dynamics of interest are photoinduced and involve valence electrons so they are typically initiated by ultrafast optical lasers. It has been demonstrated that short and intense X-ray pulses such as the ones delivered by the LCLS can be successfully employed as a probe as X-Ray induced fragmentation of the evolving system carries the signature of the ongoing reaction [1].
Experiment
The ring opening of 1,3-Cyclohexadiene (1,3-CHD) is a good example of a concrete realization of the ideas outlined above. Upon the absorption of a UV photon 1,3-CHD undergoes a complex isomerization reaction which involves the crossing of two conical intersections, as depicted schematically in figure 1a. One of the possible reaction products is the linear isomer 1,3,5-Hexatriene (1,3,5-HT) which then exhibits further conformational changes at the expenses of its vibrational energy. The time scale of the ring opening is on the order of 200 fs which is faster than the typical temporal jitter (~300 fs) between the X-rays and the UV pulse. The conformational changes are slower, on the order of 1-2 ps, so they can be reliably resolved by X-ray fragmentation. Among other experimental evidence of the ongoing dynamics a steady increase in the yield of protons when the X-ray pulse follows the UV pulse was observed (see fig. 1b). This was attributed to a general increase in the average length of C-H bonds due to the vibrational energy deposited in the system following the electronic de-excitation.
A much higher temporal resolution can be achieved if X-Ray pulses are employed both as pump and probe. A technique with which it is possible to obtain two few fs short X-Ray pulses with a delay of 2-20 fs has been proposed and recently demonstrated[2]. This scheme has paved the way to the investigation of a new class of phenomena in the field of molecular dynamics, namely the X-Ray initiated ultrafast molecular processes involving core electrons.
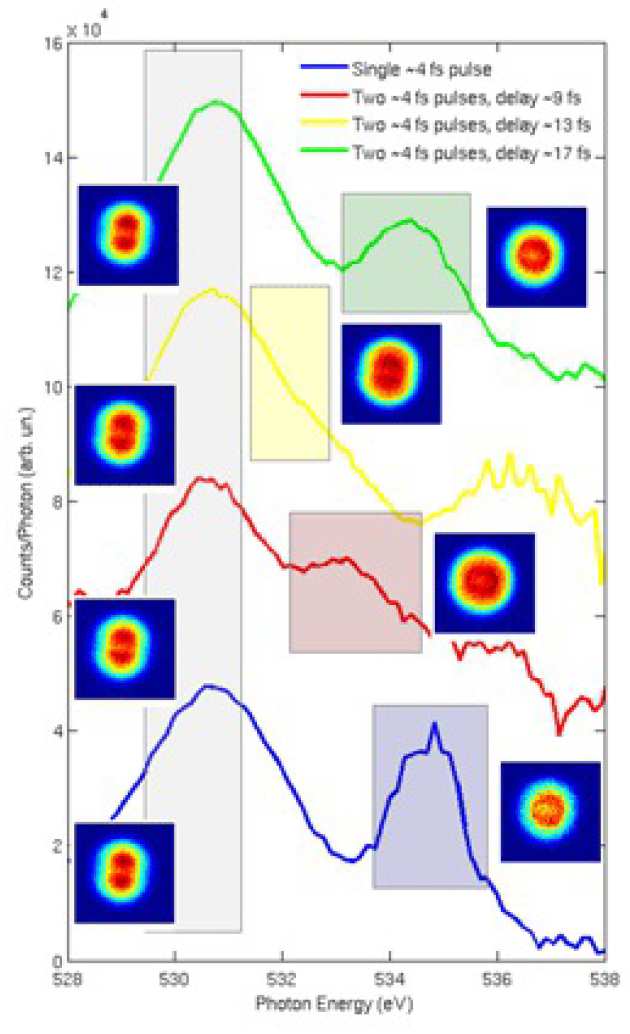 An example is provided by the ultrafast dissociation following core excitation of O2. The photon energy of the FEL was chosen to be resonant with a core excitation corresponding to the promotion of an electron from the 1σu to the 1πg* molecular orbital. Transitions involving core electrons are normally followed by Auger relaxation which leaves the ion in one of the energetically allowed valence-excited states. In case of O2 these states are not bound so dissociation follows. A second X-ray pulse probed the dissociating system by exciting further core excitations on the cation. Information about the dynamics was provided by the total ion yield,the kinetic energy spectrum and angular distribution of the atomic fragments resulting from the dissociation.
An example is provided by the ultrafast dissociation following core excitation of O2. The photon energy of the FEL was chosen to be resonant with a core excitation corresponding to the promotion of an electron from the 1σu to the 1πg* molecular orbital. Transitions involving core electrons are normally followed by Auger relaxation which leaves the ion in one of the energetically allowed valence-excited states. In case of O2 these states are not bound so dissociation follows. A second X-ray pulse probed the dissociating system by exciting further core excitations on the cation. Information about the dynamics was provided by the total ion yield,the kinetic energy spectrum and angular distribution of the atomic fragments resulting from the dissociation.
In a SASE-FEL the natural shot-to-shot variability of the physical parameters of the electron bunch can be exploited to gather further information on the system under study. Although the electron beam energy was set to give a resonant FEL wavelength equal to the 1σu - 1πg* transition each dataset contained a significant number of shots at different electron beam energies i.e. different photon energies. In fig 2 the yield of O2+ per photon is plotted as a function of photon energy for the case of a single X-Ray pulse or two pulses at variable delays. The main peak which is present in all the spectra is the 1σu – 1πg* resonance. The corresponding projection of the momentum distribution on the detector plane is shown in the form of VMI images on the left. No significant difference in the distributions can be seen as the signal is coming from molecules which interacted with only one of the two pulses.
The additiona l peak in the spectra has a rather different behaviour. Its angular distribution is close to isotropic and it exhibits a clear energy shift as a function of the delay. This is the signature of a core-excitated state that it accessible only if the molecu le undergoes the 1σu - 1πg* transition and subsequent Auger decay. As the molecule dissociates the energy difference between this state and the initial state changes according to the shape of the potential energy curve and this is ultimately reflected in the shift of the peak.
References and relevant links
[1] V. J. Petrovich, M. Siano, et al., Phys. Rev. Lett. 108, 25306 (2012).
[2] P. Emma, K. Bane, et al., Phys. Rev. Lett. 92, 074801 (2004).
Introduction
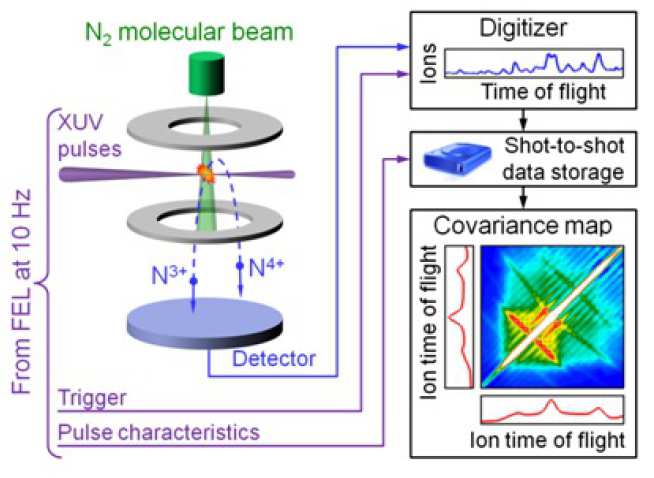
Electron dynamics in molecules can be revealed in pump-probe experiments with femtosecond and sub-femtosecond delays. In these experiments, the molecules are ionised and often fragment. The kinetic energies of photoelectrons and photoions are measured in a time-of-flight or position-sensitive spectrometer. The typical spectra are very rich, reflecting a variety of competing ionisation and fragmentation processes. These processes can be resolved using coincidence techniques that uniquely correlate photoelectrons and photoions belonging to the same process [1]. Unfortunately, coincidence experiments require low counting rates, typically much less than one event per laser shot. Such mode of operation is very inefficient in experiments where many events can be induced by a single laser shot.
To overcome the counting-rate limitation of coincidences, we developed an enhanced version of the covariance mapping technique [2]. The enhancement over the standard covariance mapping consist in using the partial covariance [3], i.e. the part of covariance that is independent of laser pulse energy fluctuat ions, which allows us to increase the counting rate substantially. With the enhancement, the experiments can be performed while thousands of molecules are ionized per laser shot, clearly precluding the possibi lity of a c o incidence analys is.
Proof of concept: Coulomb explosion of diatomic molecules
The concept of partial covariance mapping was tested in an experiment performed at the FLASH free-electron laser (FEL) in Hamburg using intense femtosecond pulses of VUV photons (see figure 1). Recording the FEL pulse energy at each shot allowed us to implement partial covariance mapping (see figure 2). The maps unravelled a detailed picture of all significant Coulomb explosion pathways, extending up to the N4+–N5+ channel for nitrogen and up to the I8+–I9+ channel for iodine. The measured ion kinetic energies are in a good agreement with classical trajectory simulations of the Coulomb explosion [4, 5]. The understanding of the explosion dynamics of diatomic molecules opens up research for more complex systems, such as C60[6].
Dynamics of Hollow Atom Formation
To demonstrate that partial covariance mapping can be used to monitor directly the electron dynamics, an experiment on atomic neon was performed at the LCLS FEL in Stanford using 1062 eV photons. At this photon energy the inner K shell becomes accessible and the high intensity induces many different ionisation sequences, such as shown in figure 3. As the various electron kinetics energies overlap, the partial covariance technique is essential to resolve the ionisation sequences (see figure 4). The variation in the branching ratios of some of these sequences as the function of the x-ray pulse duration and energy allowed us to study experimentally the dynamics of hollow atom formation on the few-femtosecond timescale [7, 8].
Theory and simulations of covariance mapping
The successful implementation of the partial covariance mapping initiated a theoretical research into other forms of this technique. We have shown [9]:
- that the covariance mapping formula automatically removes false coincidences,
- how to extend partial covariance mapping to several fluctuating parameters,
- that covariance mapping can be extended to 3 dimensions but not higher,
- the explicit formula for partial covariance mapping in 3 dimensions,
- an alternative method of contingent covariance mapping
Current researchers
L. J. Frasinski1, V. Zhaunerchyk2, M. Mucke2, R. J. Squibb1, M. Siano1, J. H. D. Eland2,3, P. Linusson4, P. v. d. Meulen4, P. Salén4, R. D. Thomas4, M. Larsson4, L. Foucar5,6, J. Ullrich5,7,8, K. Motomura9, S. Mondal9, K. Ueda9, T. Osipov10, L. Fang10, B. F. Murphy10, N. Berrah10, C. Bostedt11, J. D. Bozek11, S. Schorb11, M. Messerschmidt11, J. M. Glownia11, J. P. Cryan11, R. Coffee11, O. Takahashi12, S.Wada13, M. N. Piancastelli2, R. Richter14, K. C. Prince14, R. Feifel2, O. Kornilov15, M. Eckstein15, M. Rosenblatt15, C. P. Schulz15, A. Rouzée15, J. Klei15, A. Lübcke16, F. Schapper16, P. Johnsson17, D. M. P. Holland18, T. Schlatholter19, T. Marchenko20, S. Düsterer21, M. J. J. Vrakking15
1Blackett Laboratory, Imperial College London, SW7 2AZ, United Kingdom
2Department of Physics and Astronomy, Uppsala University, SE-751 20 Uppsala, Sweden
3Physical and Theoretical Chemistry Laboratory, Oxford University, OX1 3QZ, UK
4Department of Physics, Stockholm Unive rsity, SE-106 91 Stockholm, Sweden
5Advanced Study Group of the Max Planck Society, 22607 Hamburg, Germany
6Max Planck Institut für medizinische Forschung, 69120 Heidelberg, Germany
7Max Planck-Institut für Kernphysik, 69117 Heidelberg, Germany
8Physikalisch-Technische Bundesanstalt, 38116 Braunschweig, Germany
9Institute for Interdisciplinary Material Research, Tohoku University, Sendai 980-8577, Japan
10Department of Physics, Western Michigan University, Kalamazoo, MI 49008, USA
11SLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA
12Department of Chemistry, Hiroshima University, Higashi-Hiroshima 739-8526, Japan
13Department of Physical Science, Hiroshima Univ., Higashi-Hiroshima 739-8526, Japan
14Elettra-Sincrotrone Trieste, 34149 Trieste, Italy
15Max-Born Institut, Max-Born Strasse 2A, 12489 Berlin, Germany
16Ecole Polytechnique Fédérale de Lausanne, CH- Lausanne, Switzerland
17Department of Physics, Lund University, 22100, Lund, Sweden
18STFC Daresbury Laboratory, Warrington, Cheshire WA4 4AD, Unit ed Kingdom
19University of Groningen, NL-9747 AA Groningen, the Netherlands
20Laboratoire de Chimie Physique-Matière et Rayonnement, CNRS and UPMC, 75231 Paris, France
21Deutsches Elektronen-Synchrotron (DESY), D-22603 Hamburg, Germany
References and relevant links
[1] J. H. D. Eland, Adv. Chem. Phys. 141, 103−563 (2009).
[2] L. J. Frasinski, K. Codling, and P. A. Hatherly, Science 246, 1029−1031 (1989).
[3] L. J. Frasinski, et al, J. Electron Spectrosc. Relat. Phenom. 79, 367–371 (1996).
[4] O. Kornilov, et al, J. Phys. B: At. Mol. Opt. Phys., 46, 164028 (2013), open access.
[5] Europhysics News, 45, no. 1.
[6] B. F. Murphy, et al, Nature Communications (submitted January 2014).
[7] L. J. Frasinski, et al, Phys. Rev. Lett. 111, 073002 (2013), open access.
[8] V. Zhaunerchyk, et al, J. Phys. B: At. Mol. Opt. Phys., 46, 164034 (2013).
[9] V. Zhaunerchyk, et al, Phys. Rev. A (submitted November 2013).
Introduction
Access to single attosecond pulses in the XUV and the VUV energy ranges, on the same experimental setup [1], offers the opportunity to trigger and probe ultrafast multi-electronic processes on systems such as hole migration in organic molecules. The complexity of ionization and fragmentation channels following the pump interaction with those systems make necessary to record ions and photo-electrons simultaneously with high sensitivity.
The main goal of the project is the real time measurement of charge migration on Glycine through the sp-LEAD (single photon Laser Enabled Auger Decay) mechanism [2]. To achieve this a Velocity Map Imaging Spectrometer (VMIS) [3] to record electron kinetic energy spectrum and an ion Time Of Flight (iTOF) have been designed and constructed.
Experiment
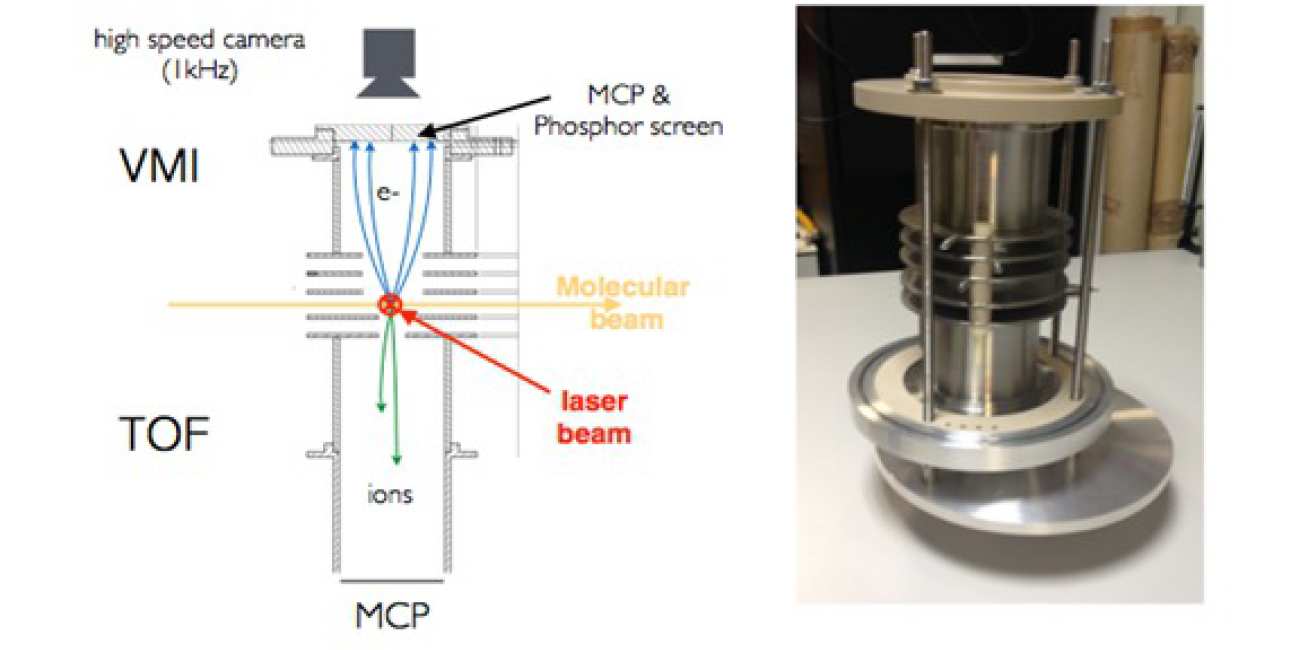
A VMI-TOF coupled spectrometer has the advantage to be compact (figure 1) and therefore can be introduced in an attosecond pump-probe beam line with minimal constraints.
Photo-electron measurement is done by the VMIS which is able to detect photo-electrons with kinetic energies up to 90 eV with a resolution of a few percent (figure 2-a). All the electrons emitted from the interaction region are guided by the electro-static field to the MCP detector, the collection solid angle is 4π. A high speed camera records images at a 1kHz repetition rate and is synchronized with the laser. Each frame is treated and saved in real time. This ensures single shotmeasurements on the VMI side.
Ion fragments are detected by an iTOF in a Whiley-Maclaren [4] arrangement designed to obtain a resolution of 100 amu. Figure 2-b shows simulated TOF of systems separated by 1 amuaround 81 amu. This is enough to resolve ions from a molecule like glycine (75 amu) but can be pushed to bigger masses if needed. The TOF is connected to a digitizer to achieve 1kHz resolved measurements.
The single shot recording of photo-electrons and ions allows covariance mapping [5] to be used for fine data analysis and will link precisely every fragment signal to its related electron signal.
Current Researchers
D.J. Walke, D. Fabris, P. Matia-Hernando, T. Barillot, T. Witting, W. Okell, L. J. Frasinski and J. P. Marangos, J.W.G. Tisch.
References and relevant links
[1] D. Fabris et al., arXiv:1311.4738, (2013).
[2] B. Cooper and V. Averbukh, Physical Review Letters 111, 8 (2013).
[3] Eppink et al., Rev. Sci. Instruments 68, 9 (1997).
[4] W. C. Wiley and I. H. McLaren, Rev. Sci. Instruments 26, 12 (2004).
[5] L. J. Frasinski et al., Science 246, 4933 (1989).
Introduction
Excitation of a coherent superposition of electronic states initiates electronic dynamics in molecules. Observing and controlling the effects of the coupled electron-nuclear dynamics is a target of attosecond spectroscopy.
In order to study such effects computationally we have implemented the Ehrenfest method and approximate gradient and Hessian within a CASSCF formalism. Using this approach, we can study the evolution of a non-stationary electronic wavefunction for fixed atomic nuclei, and where the nuclei are allowed to move, to investigate the differences resulting from nuclear motion for the first time (figure 1).
Theory
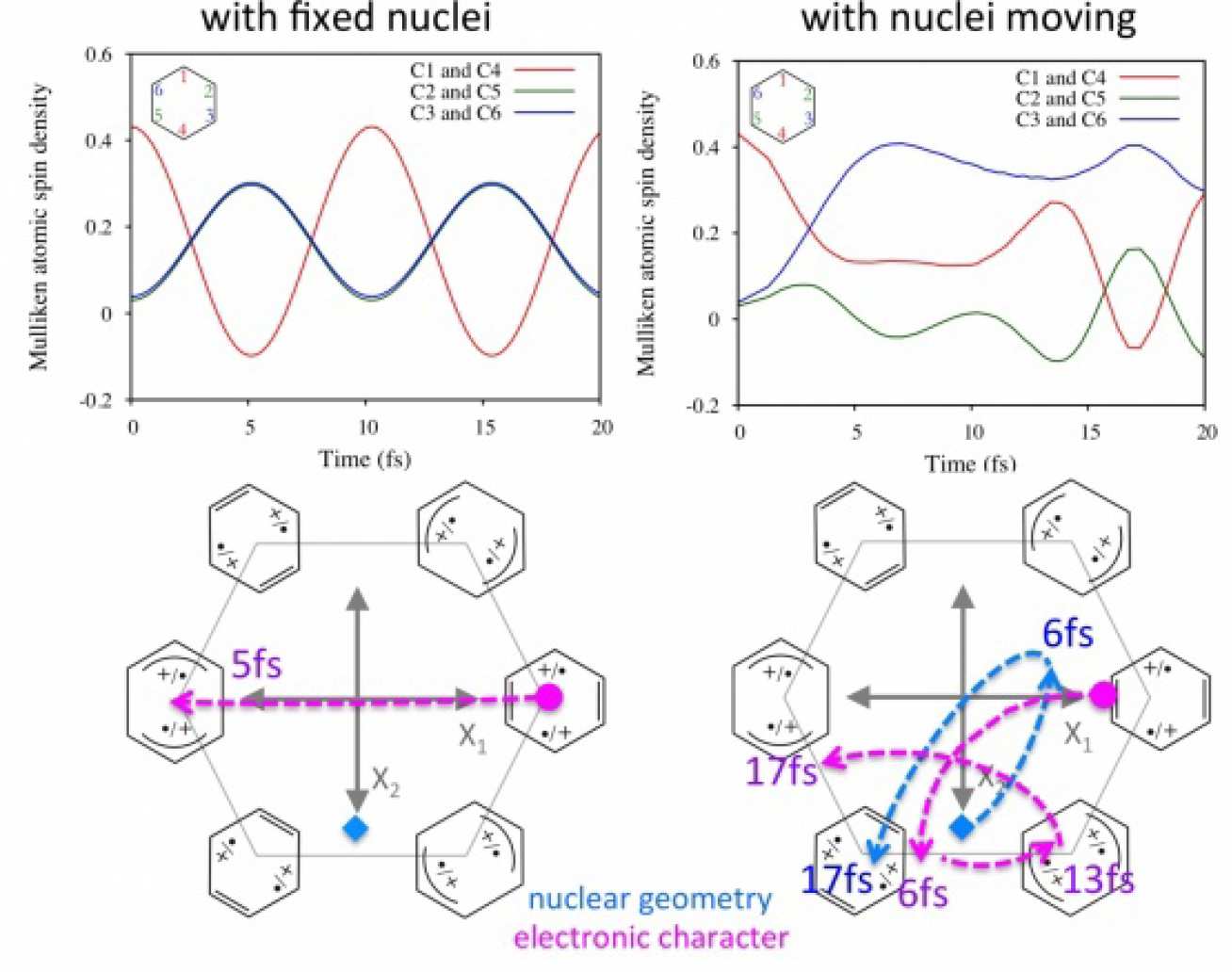
The method is general and can be used to study any coupled electron-nuclear dynamics with a CASSCF wavefunction. Our initial results [1-2], suggest two limiting mechanisms: nuclear motion may occur very quickly as a result of the electron dynamics1; alternatively, rapid oscillating electronic charge migration have little direct effect on nuclear motion.
Two further conceptual ideas have developed from these studies using Ehrenfest dynamics. Firstly, it appears that initial electron dynamics can be effectively engineered in the avoided crossing region close to a conical intersection. Secondly, it appears that one can control the nuclear dynamics by controlling the amplitudes and phases of the initial superposition of the populated electronic states of the cation. For example, changing the real phase allows us to determine which minimum on the ground state potential energy surface we decay towards.
Current researchers
M. Vacher, J. Meisner, D. Mendive-Tapia, M. J. Bearpark, M. A. Robb
References and relevant links
[1] D. Mendive-Tapia et al., J. Chem. Phys. 139, 044110 (2013).
[2] M. Vacher, D. Mendive-Tapia, M. J. Bearpark, and M. A. Robb Theoretical Chemistry Accounts, submitted.
Introduction
Ultrafast charge migration, following sudden ionisation or excitation, is believed to be a universal response of extended molecules and is likely also to occur in some form in many condensed phase systems [1, 2]. This process occurs as a result of electron correlation, and is predicted to occur on the few femtosecond to sub-femtosecond timescales. Using the molecule glycine, we show that the dynamics resulting from sudden ionisation in the inner valence region evolve sufficiently slowly that they are able to be measured using X-ray pulses from free electron laser facilities.
Proposed experiment
The proposed pump-probe scheme (seen in Fig. 1) uses a pump pulse of 275 eV (below the K edge of carbon) that will ionise from the inner valence region. A second probe pulse of the same energy can excite an electron from the core shell to the inner valence region. By measuring the Auger electrons produced by the subsequent refilling of the core hole, the dynamics occurring in the inner valence region can be tracked.
As an example for this general scheme we choose to investigate the glycine molecule. The three most abundant conformers of glycine are commonly referred to as Gly I, Gly II and Gly III. To obtain glycine in the gas phase, the sample will be heated in an oven with operation range ∼150–210 ◦ C where we expect the Gly I and Gly III conformers to dominate the sample.
We are interested in the dynamics that result from ionising from the inner valence region, specifically from the 10A’ orbital of glycine. Sudden ionisation from this orbital results in a superposition of two eigenstates, with roughly 50% contribution from a single hole (1h) and 50% contribution from two-hole one-particle configurations (2h1p). The resulting dynamics arise purely from electron correlation. The hole survival probability will oscillate in time as the two eigenstates evolve with different phases as they have different energies. Nuclear dynamics will lead to a significant distortion of the molecular geometry (around 20 fs) leading to damping or to modifications of the oscillations.
Theory
Our calculations[3] using ADC(2)x for the hole survival probability, S(t), for ionisation from the 10A’ orbital of glycine are shown in Fig. 2. These show the survival probability for the Gly I conformer, the Gly III conformer and a 2:1 ratio of both Gly I and Gly III which we expect from the operational temperature range (150–170° C). The dynamics are sufficiently slow to be accessible to measurement using the temporal resolution available from e.g. the LCLS X-ray FEL located at SLAC. Fig. 2 illustrates the robustness of the measurement by simulating the effect of using an X-ray pulse of different durations, 2 fs, 4 fs and 6 fs, whereas we expect the pulse duration to be nearer 3 fs.
Current researchers
B. Cooper, P. Kolorenc, L. J. Frasinki, V. Averbukh and J. P. Marangos
References and relevant links
[1] J. Breidbach and L. S. Cederbaum, J. Chem. Phys. 118, 3983 (2003).
[2] F. Remacle and R. D. Levine, Proc. Nat. Acad. Sciences 103, 6793 (2006).
[3] B. Cooper, et al., Faraday Discuss 171 (2014) accepted.
Introduction
An instant ionization of a molecule may lead to the population of several cation states, having different charge distributions. This leads to the migration of the hole (positive charge) density between the configurations corresponding these cation states. The period of the migration is dependent on the difference of the cation state energies, ∆E. If only two states are involved in the process, the period of the migration will be T = 2π/∆E. Usually the charge migration process is studied neglecting the nuclear dynamics, i.e. freezing the nuclei at the equilibrium positions of the neutral molecule. In the case of ultrafast nuclear dynamics latter may change the picture of electronic dynamics significantly. We analyze the effect of nuclear dynamics on charge migration process in Glycine (NH 2 -CH 2 -COOH).
Theory
The analysis of the nuclear dynamics can be split into two parts. First, the calculation of the potential energy surfaces (PES) for the cation states populated after an instant ionization. In the second step a quantum mechanical analysis of the relaxation process by the nuclear wavepacket propagation on the obtained electronic states (PES) will be performed. Potential energy surfaces for the cation states are calculated using the Algebraic-Diagrammatic Construction (ADC) method [1], which gives the ionization energies at a given configuration of the nuclei. The propagation of the nuclear wavepacket will be performed by Multi-Configuration-Time-Dependent-Hartree method [2].
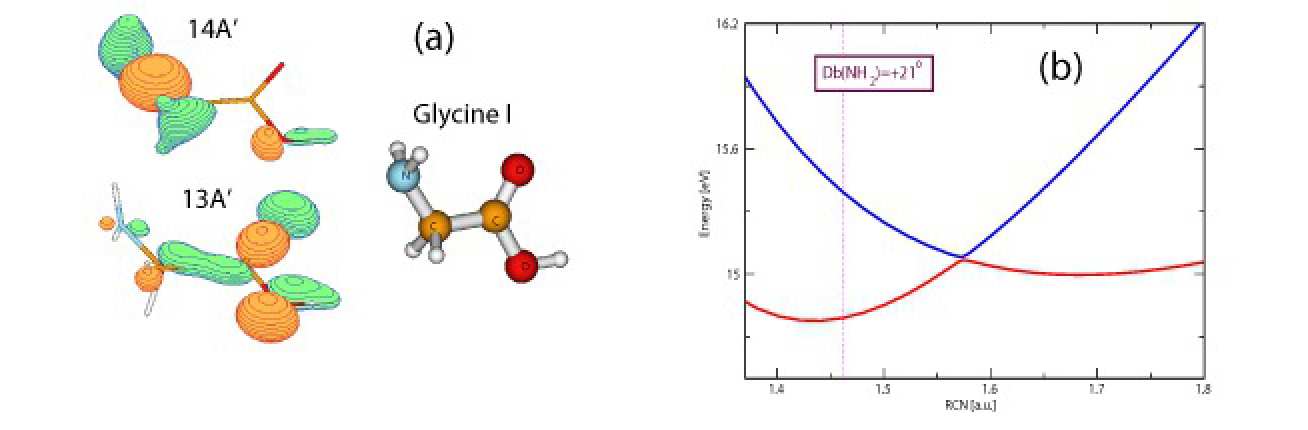
Charge migration in Glycine was analyzed theoretically for the fixed nuclear configuration [3]. For our analysis we initially populate two cation, 4-th and 5-th excited, states, which mainly represent a superposition of two single-hole orbitals, 13A ′ and 14A ′ , shown in Fig. 1 (a). The energy difference of about 0.6 eV leads to the migration of the charge with a period of 7 fs when the nuclear motion is not present. Due to the large dimensionality of the Glycine system (totally 24 degrees of freedom), we have to drop some degrees of freedom, choosing the most important coordinates which take part in the relaxation process. Our analysis shows a very important role of the NH 2 binding coordinates and negligible role of the CH 2 binding coordinates. An important point is the conservation of the C s group symmetry present for the ground state of the Glycine neutral. The energy points are calculated using the McLean-Chandler 6-311G(2df,2p) basis set.
The nuclear dynamics may be more pronounced when the cation PES exhibit conical intersections. Fig. 1 (b) shows the presence of a conical intersection between the cation couple along the C-NH 2 binding coordinate changing one of the NH 2 angles. Here a very fast transition to the lower electronic state occurs, followed by the further movement of the nuclear wavepacket towards the lower energies. In the reality the cation states we have chosen exhibit many conical intersections. We estimate for the time scale of the nuclear dynamics to be in femtosecond range, which leads to the big influence of the nuclear motion on charge migration process.
Current researchers
S. Sukiasyan, V. Zakjevskii, J. P. Marangos and V. Averbukh
References and relevant links
[1] J. Schirmer, L.S. Cederbaum and O. Walter, Phys. Rev. A 28, 1237 (1983).
[2] M. H. Beck, A. J¨ ackle, G. A. Worth, and H.-D. Meyer, Phys. Rep. 324, 1 (2000).
[3] A.I. Kuleff, J.Breidbach and L.S. Cederbaum, J. Chem. Phys. 123, 044111, (2005).
Introduction
Here we investigate single-photon Laser Enabled Interatomic Coulombic Decay- spLEICD, in rare gas heteroatomic clusters such as NeAr. Interatomic Coulombic Decay- ICD for short, is a non-radiative decay process which takes place in atomic (or molecular) clusters with an inner-valence vacancy on one atom [1]. This vacancy is not energetic enough to cause local second-ionization, as in auger decay [2]. However being part of a cluster gives it the opportunity to induce non-local second-ionization of the whole cluster, as this requires less energy. This process involves relaxation of an outer-valence electron on the singly ionized atom into the vacancy, while an outer-valence electron on a neighbouring atom is ejected into the continuum as a result of Coulomb interaction between electrons.
In NeAr however, with an Ar inner-valence vacancy, the 3p → 3s transition on Ar is not energetic enough to ionize Ne. Therefore ICD cannot proceed unless the system is injected with an additional amount of energy by an external source, such as a photon. This process is called single-photon Laser Enabled ICD (spLEICD) (Fig. 1), here investigated for the first time, and builds upon the recent work by Cooper and Averbukh on single-photon Laser Enabled Auger Decay (spLEAD) [3].
Theory
The quantity we use in order to characterize spLEICD is the photo-ionization cross-section, σ, a measure of the likelihood for spLEICD to take place. The cross-section is obtained using Stieltjes imaging technique applied to the extended second-order Algebraic Diagrammatic Construction [ADC(2)x] transition matrix elements. For NeAr[3s−1], σ spans a photon energy range of 10 − 17 eV; beyond this range spLEAD takes over (Fig.2 a).
We explore how the spLEICD cross-section varies with internuclear distance. Fig.2 b is a numerical ap- proach to this problem. It exhibits two features that represent a R-12 behaviour for R = 2.5 − 3.9 Å, and a R-6 behaviour for R = 4 − 5.2 Å. This shows that the total spLEICD cross-section, in contrast to the conventional photoionization, is very strongly dependent on the internuclear distance.
Current researchers
L. Bahmanpour, B. Cooper and V. Averbukh.
References and relevant links
[1] L.Cederbaum, J. Zobeley, F. Tarantelli, Phys. Rev. Lett. 79, 4778 (1997).
[2] B. Crasemann, ”Atomic Inner-Shell Physics”, (1985).
[3] B. Cooper, V. Averbukh, Phys. Rev. Lett. 111, 083004 (2013).
[4] V. Averbukh, P. Kolorenc, Phys. Rev. Lett. 103, 183001 (2009).