resume

PhD Theoretical Physics, Menéndez Pelayo International University, Spain (advisors: J.F. Lutsko and F. Otálora). Included placement in Université libre de Bruxelles, Belgium; MS Crystallography and Crystallisation, Menéndez Pelayo International University, Spain; MS Medical Physics, University of Seville, Spain; Lic. (5 yrs degree) Physics, University of Seville, Spain.
About
Miguel is now a Visiting Research Associate at the Department of Chemical Engineering, Imperial College, co-founder of Vortico Tech consulting (link) and Financial and Machine Learning Consultant. Prior to this he was Assistant Professor in Computer Science & Machine Learning at Comillas Pontifical University - ICAI, Madrid, with his ICAI web page available here.
I was a research associate in the Complex Multiscale Systems group in the Department of Chemical Engineering at Imperial College London, headed by Prof. Serafim Kalliadasis. My PhD research was on non-classical nucleation theories, being located in both Granada (Spain) and in Brussels (Belgium), supervised by James F. Lutsko and F. Otálora. Prior to this I did a Masters degree in Medical Physics (University of Seville) and another one in Crystallography and Crystallization (University of Menéndez-Pelayo). To complement my Physics studies, I also completed three years of a separate degree in Mathematics at University of Seville (Spain).
email: m.duran-olivencia@imperial.ac.uk
Research
Thus far my research has been focused on non-equilibrium phenomena. Developing mesoscopic models for these kind of processes plays a prominent role when the interest lies on obtaining tractable theoretical expressions to bypass both classical thermodynamics and simulation-based methods. In this regard, classical Density Functional Theory (DFT) has been widely used as the perfect balance between both developments. This framework enables the consideration of molecular-level detail in the calculations, whilst being computationally tractable, when compared to molecular simulations. An extension to the non-equilibrium scenario, dynamical density functional theory (DDFT), has more recently been pursued.
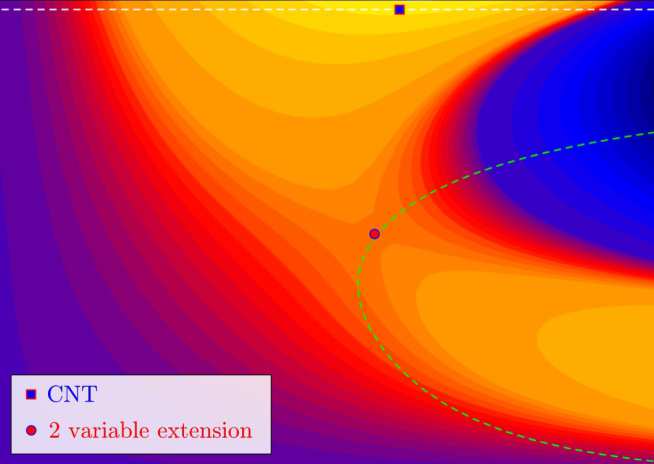
Over the course of the last decades, there has been an explosion of interest in generalising DDFT to describe more general systems or random processes, e.g. to account for colloidal fluids with inertia and hydrodynamic interactions, or to describe nucleation in a self-consistent fashion. In the case of nucleation, DDFT is hardly applicable given the stochastic nature of the process. Notwithstanding, it has been recently shown that such a paradigm can be extended to describe density fluctuations in colloidal fluids [Lutsko, J. Chem. Phys. 136 034509 (2012)]. Based on this fluctuating-hydrodynamic picture (which in turn may be simplified to DDFT when averaged over the noise) a multi-parameter theory of nucleation has been developed. Applied to simple parametrised density profiles, the theory is able to reproduce key results from the classical paradigm, whilst correcting many of its inconsistencies and drawbacks. Additionally, fluctuating-hydrodynamics has afforded great flexibility when compared to the classical model, such as being able to investigate the capillary model for clusters. Combined with DFT to prescribe the work of cluster formation, the so-called Mesoscopic Nucleation Theory shed light both on how the confinement can affect nucleation and on the multi-step nature of the process (see figures below).
My current research is focused on trying to extend the unified DDFT for colloidal fluids, which includes inertia and hydrodynamic interaction [Goddard et al., J. Phys.: Condens. Matter 25 035101 (2013)], to complex fluids in relation to my prior research and for other varied applications.
Selected publications
- M.A. Duran-Olivencia, B.D. Goddard and S. Kalliadasis. Dynamical density functional theory for orientable colloids including inertia and hydrodynamic interactions, Journal of Statistical Physics 164(4), 785-809 (2016) - (arXiv)
- J.F. Lutsko and M.A. Duran-Olivencia. A two-parameter extension of classical nucleation theory, Journal of Physics: Condensed Matter 27(23), 235101 (2015)
- M.A. Duran-Olivencia and J.F. Lutsko. Mesoscopic nucleation theory for confined systems: A one-parameter model, Physical Review E 91, 022402 (2015)
- M. Sleutel, J.F. Lutsko, A.E.S Van Driessche, M.A. Duran-Olivencia and Dominique Maes. Observing classical nucleation theory at work by monitoring phase transitions with molecular precision, Nature Communications 5, 5598 (2014)
- J.F. Lutsko and M.A. Duran-Olivencia. Classical nucleation theory from a dynamical approach to nucleation, Journal of Chemical Physics 138, 244908 (2013)