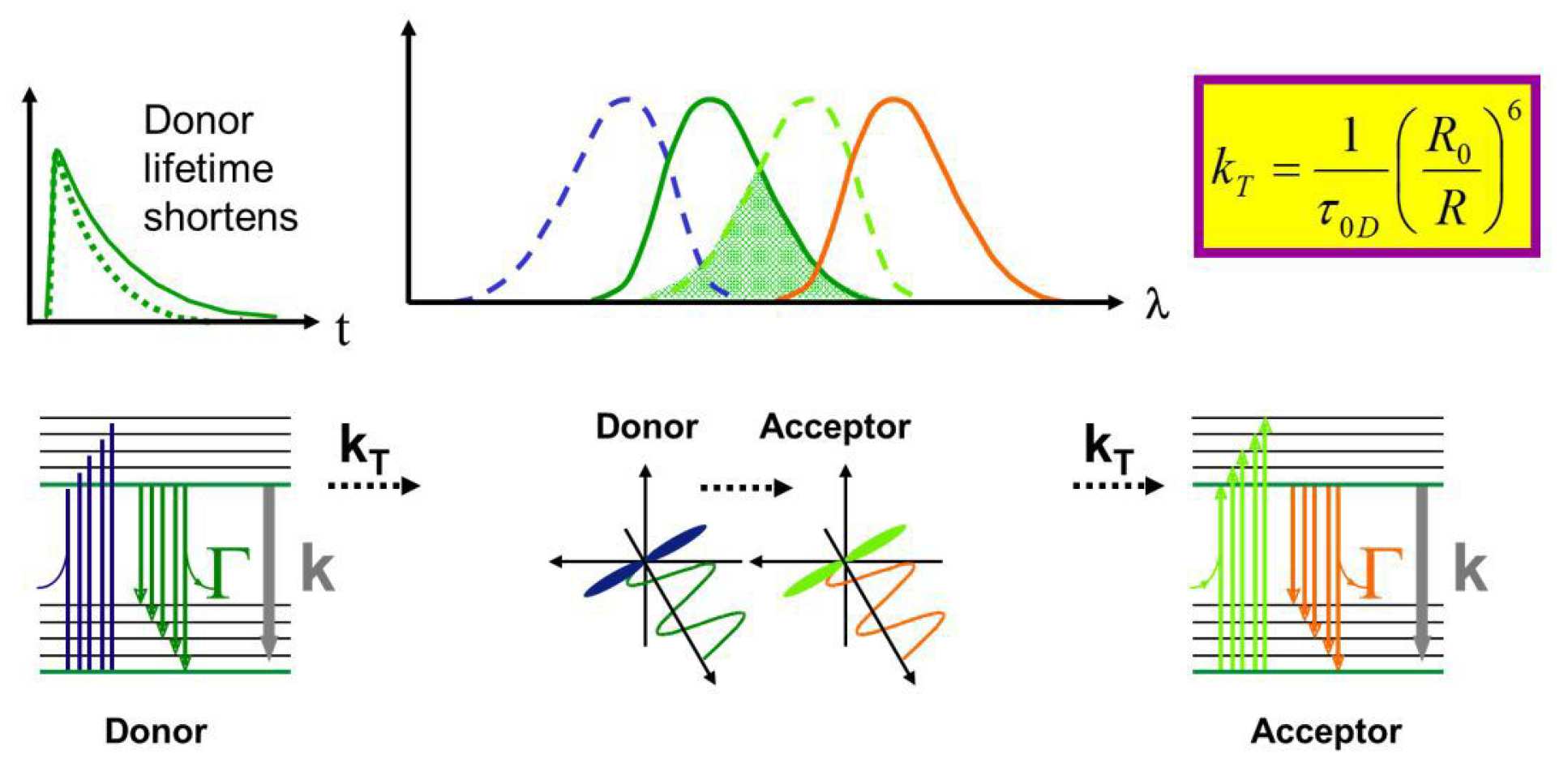
A key application of fluorescence microscopy is to study molecular biological processes in cells and tissue, for which it is important to be able to map the spatio-temporal organisation of protein interactions. By using multiple fluorescent labels that can be distinguished by an appropriate spectroscopic parameter, it is possible to image the colocalisation of different proteins but this is only possible to the precision of the imaging system, for which the resolution is usually limited to a few 100 nm by diffraction. It is, however, possible to obtain information about the distance between pairs of appropriate fluorophores on a scale of nm by studying the resonant transfer of energy between them by exploiting spectroscopic measurements of fluorescence. This energy transfer was first described by Förster1 and Förster resonant energy transfer (FRET)2 provides both a means to detect protein interactions and a spectroscopic ruler3 to measure the distance between fluorophores. The energy transfer quenches the fluorescence of a particular fluorophore (the donor) when the second fluorophore (the acceptor) is in close proximity – typically less than ~10 nm. The quenching arises from an interaction of the donor and acceptor dipoles via a non-radiative transfer of energy from an excited donor fluorophore to a ground state acceptor that requires an overlap between the emission spectrum of the donor and the excitation spectrum of the acceptor4, and also requires that the transition dipole moments of the donor and acceptor are not perpendicular5. The FRET efficiency, kT, varies with the inverse sixth power of the distance between donor and acceptor and is usually negligible beyond ~10 nm. Thus the observation of FRET between fluorescently labelled proteins (or other biomolecules) provides strong evidence of their interaction. Furthermore, with measurements of sufficient signal to noise ratio, the FRET efficiency can be used to determine the distance between “FRETing” fluorophores, which can be used to study protein structure and conformational changes of multi-labelled molecules. Fluorescence imaging techniques that can measure FRET in each pixel can thus provide a method to map protein colocalisation, interactions and conformational changes in time and space. FRET can occur between different molecular species (“heteroFRET”) or between the same fluorophore species (“homoFRET”) where there is overlap between the excitation and emission spectra.
FRET is is widely employed to detect and monitor protein-protein interactions, either as an end point in assays of fixed cells or as a dynamic event in live cells. In fixed cells, the proteins of interest may be labelled using antibodies or using dyes that directly bind their target proteins. In live cells FRET can be used with genetically-expressed donor and acceptor fluorophores (such as CFP with YFP or GFP with mCherryFP) and is a powerful means to study dynamics protein interactions with single cell resolution.
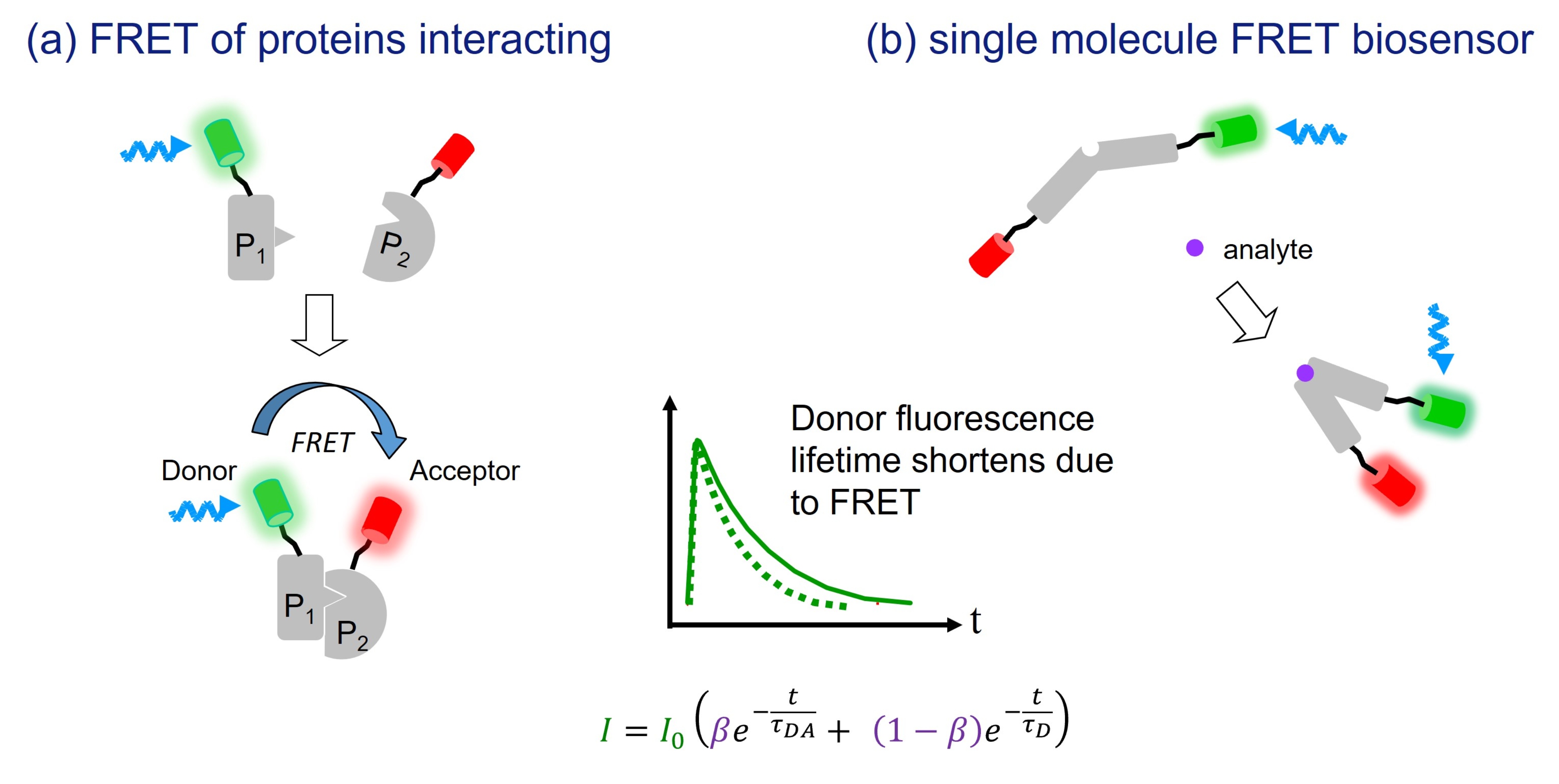
FRET can also be utilised in a range of genetically expressed single molecule FRET biosensors, of which the “Cameleon” calcium sensor6 is perhaps the best known. These biosensors incorporate both the donor and acceptor fluorophore in a single molecular construct and change their conformation (and therefore the FRET efficiency) when they bind to their analyte. A wide array of intramolecular FRET biosensors have now been reported, reading out e.g. calcium7, potassium8, chloride9, GTP10, IP311, PIP212, and others13. In addition, there are “cleavable” sensors for which the FRET signal disappears upon activation, such as a FRET sensor for monitoring calpain proteolytic activity14, to further complement the wide range of intermolecular FRET readouts of protein interactions.
Imaging FRET is a powerful approach to study the spatio-temporal organisation of, e.g. cell signalling processes for the study of disease mechanisms. Unfortunately, detecting and quantifying the resonant energy transfer is often not straightforward. Although FRET can be observed via fluorescence intensity imaging, e.g. of the acceptor fluorescence when the donor is excited, this is susceptible to a number of artefacts, such as direct excitation of the acceptor and spectral “bleed-through” of the donor fluorescence into the acceptor detection channel. Such artefacts should be minimised though careful control measurements and correction calculations15 but this is not always practical and, increasingly, other fluorescence parameters are being utilised to improve the reliability of FRET experiments including fluorescence spectra, lifetime and polarisation anistropy.
FLIM is emerging as one of the most robust approaches to read out FRET, e.g.16, 17, 18, because the donor fluorescence lifetime is largely independent of factors such as fluorophore concentration, excitation and detection efficiency, inner filter and multiple scattering effects, which can complicate or degrade absolute intensity measurements. In particular, the insensitivity of fluorescence lifetime measurements to fluorophore concentration and spectral cross-talk make FLIM the preferred technique when imaging samples for which the precise stoichiometry of the donor and acceptor species is unknown – as is the case when monitoring interactions between separately labelled proteins (e.g. binding partners, enzyme/substrate pairs etc.). A further advantage of FLIM applied to FRET is that the measured fluorescence decay profiles can be fitted to a double exponential decay profile and the relative population fractions of FRETing and nonFRETing donor fluorophores can be determined (and therefore the population fraction of interacting donor-labelled proteins).
We are developing a range of fluorescence-based instrumentation to utilise FRET readouts of cell signalling processes. These include our openFLIM-HCA platform for screening protein interactions and our FLIM endomicroscope and FLIM OPT platforms for in vivo preclinical FRET studies.
1 Förster, T., Annalen der Physik, 2 (1948) 55
2 Clegg, R. M., Curr Opin Biotechnol, 6 (1995) 103
3 Stryer, L. and Haugland, R. P., Proc Natl Acad Sci U S A, 58 (1967) 719
4 Haugland, R. P., Yguerabide, J. and Stryer, L., Proc Natl Acad Sci U S A, 63 (1969) 23
5 Eisinger, J. and Dale, R. E., J Mol Biol, 84 (1974) 643
6 Miyawaki, A., Llopis, J., Heim, R., Mccaffery, J. M., Adams, J. A., Ikurak, M. and Tsien, R. Y., Nature, 388 (1997) 882
7 Mank, M., Reiff, D. F., Heim, N., Friedrich, M. W., Borst, A. and Griesbeck, O., Biophysical Journal., 90 (2006) 1790
8 Ueyama, H., Takagi, M. and Takenaka S., Journal of the American Chemical Society, 124 (2002) 14286
9 Kuner, T. and Augustine, G. J., Neuron., 272 (2000) 447
10 Mochizuki, N., Yamashita, S., Kurokawa, K., Ohba, Y., Nagai, T, Miyawaki, A. and Matsuda, M., Nature, 411 (2001) 1065
11 Tanimura, Nezu, A., Morita, T., Turner, J. and Tojyo, Y., Journal of Biological Chemistry, 279 (2004) 38095
12 Nishioka, T., Aoki, K., Hikake, K., Yoshizaki, H., Kiyokawa, E. and Matsuda, M., Mol Biol Cell, 19 (2008) 4213
13 Li, I. T., Pham, E. and Truong, K., Biotechnology letters, 28 (2006) 1971
14 Stockholm, D., Bartoli, M., Sillon, G., Bourg, N., Davoust, J. and Richard, I., Journal of Molecular Biology, 346 (2005) 215
15 Gordon, G. W., Berry, G., Liang, X. H., Levine, B. and Herman, B., Biophysical Journal, 74 (1998) 2702
16 Bastiaens, P. I. and Squire, A., Trends in Cell Biology, 9 (1999) 48
17 Jares-Erijman, E. A. and Jovin, T. M., Nature Biotechnology, 21 (2003) 1387
18 Suhling, K., French, P. M. W. and Phillips, D., Photochemical & Photobiological Sciences, 4 (2005) 13