Optically sectioning microscopes
- Confocal laser scanning
- Multiphoton laser scanning
- Wide-field optical sectioning
- Light-sheet microscopy
- Total internal reflection
Confocal laser scanning fluorescence microscopy
While the conventional wide-field microscope can provide high quality images of thin samples with a (diffraction-limited) lateral resolution of ~λ/(2NA), its performance is degraded when imaging thick samples. This is because the in-focus image of the sample in the focal plane of the objective can be swamped by a diffuse background formed by light from above and below the focal plane that will also be detected but will be out of focus. In fact, a wide-field microscope collects a similar number of photons from each focal plane – the photons collected from within the depth of field of the objective lens will form a clear in-focus image and the other photons contribute to the unwanted background. This problem was overcome in 1961 by the invention of the confocal microscope1, in which a point source of light is imaged to illuminate a spot in the sample plane and the reflected or emitted light from this point is then imaged onto a pinhole in front of the detector, as depicted in the figure below. This confocal arrangement means that only light from this spot in the objective focal plane is efficiently transmitted through the pinhole to be detected, since light arriving at the detector from points at other depths in the sample will not be in focus at the pinhole. By transversely scanning the point illumination across focal plane of the objective, an optically sectioned or depth-resolved 2-D image at a single depth in the sample is obtained2. By scanning the sample through the objective’s focal plane or by moving the microscope objective, a stack of such optically sectioned images corresponding to different depths in the sample can be acquired and combined to reconstruct a high resolution 3-D image. Confocal microscopy can be implemented with reflected or fluorescence light and is usually implemented using a laser beam to provide the required point illumination. This is an example of an optical sectioning microscope and provides axial resolution, forming an image only from photons originating within the depth of field of the objective lens.
Theoretically, the lateral resolution of a confocal microscope (with an infinitely small pinhole) is approximately √2 smaller than that of the corresponding conventional (wide-field) microscope. In practice, however, this lateral resolution is not obtained using a pinhole sufficiently wide to provide sufficient signal for most applications. The axial resolution, however, is preserved in real confocal microscopes with pinhole diameters up to ~1 Airy unit (AU, where 1 AU = 1.22λ/NA) and is approximately3: λ/[n - √(n2-NA2)].
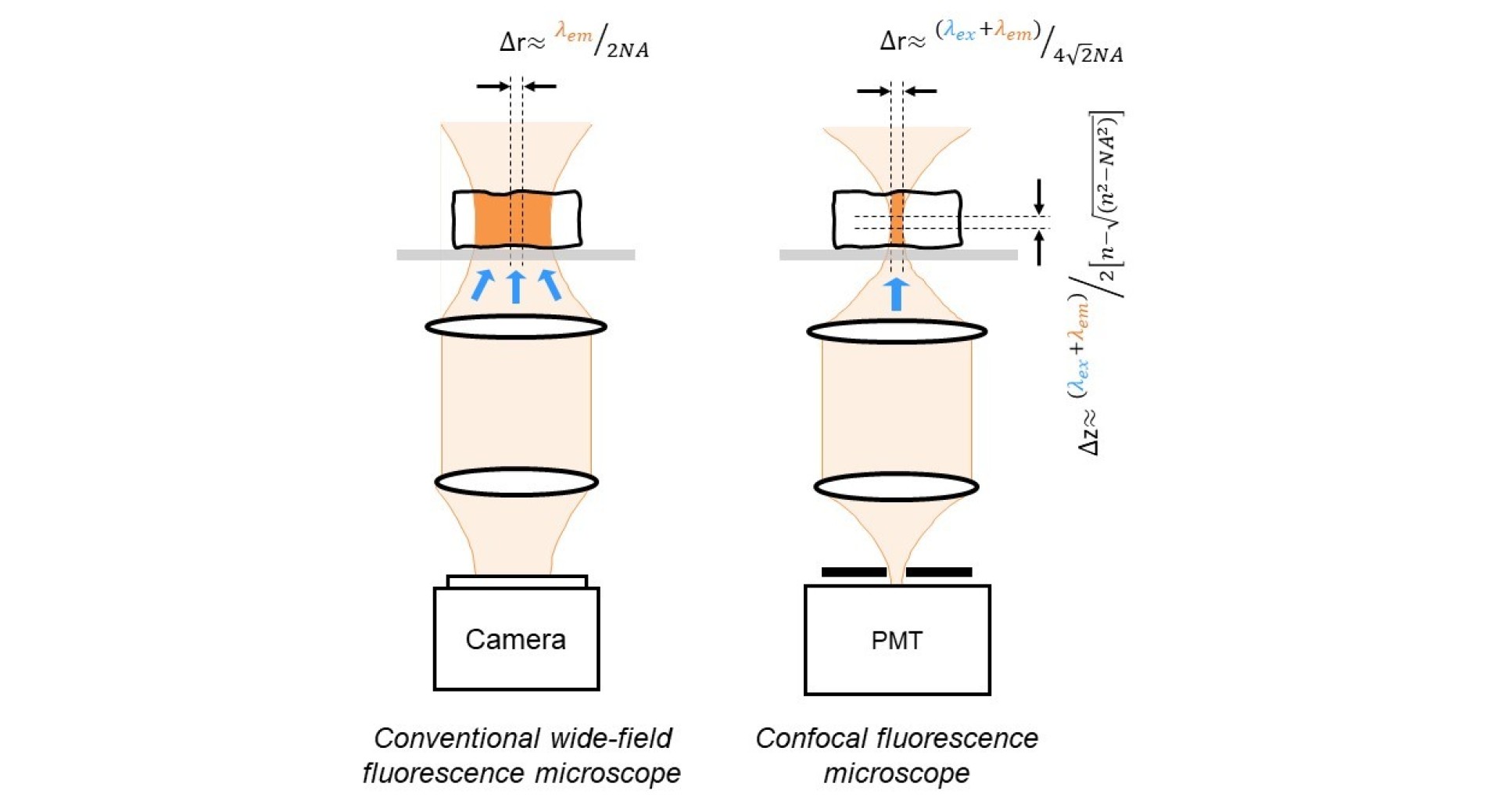
Although 3-D imaging is an attractive capability, the impact of confocal microscopes has been much wider. The elimination of the out-of-focus blur improves the image contrast and makes confocal microscopy particularly useful for quantitative imaging, which has led it to become an almost standard tool in cell biology. Furthermore, before the development of the CCD camera, the laser scanning confocal microscope was the primary means to acquire digital images and the parallel development of personal computers led to the powerful image processing capabilities that are taken for granted today – indeed, digital image processing underpins most of the current advances in all imaging capabilities.
1 Minsky, M., Scanning, 10 (1988) 128
2 Conchello, J.-A. and Lichtman, J. W., Nature Methods, 2 (2005) 920
3 Wilson T., Journal of Microscopy 244 (2011) 113
Multiphoton laser scanning fluorescence microscopy
Laser scanning confocal microscopy is routinely employed using reflected (backscattered) light or fluorescence to acquire 3-D spectroscopic images of biological samples and is becoming increasingly important for medical applications. Imaging in biological tissue, however, is strongly compromised by the strong scattering experienced by visible radiation, as well as by absorption and it is difficult to obtain high (diffraction-limited) resolution at depths greater than ~100 mm. Both scattering and absorption in biological tissue decrease significantly for longer wavelength radiation in the near infrared (NIR) but most commonly used fluorophores require visible excitation and endogenous fluorophores of interest are best excited using blue or ultraviolet (UV) radiation. An elegant approach to extend the depth for high resolution optical imaging in biological tissue is multiphoton microscopy1, which exploits nonlinear optics and uses an ultrafast laser to excite fluorescence in the sample with trains of ultrashort optical pulses that exhibit high peak powers at relatively low average powers levels. The corresponding high intensities can be sufficient for two-photon absorption, in which a molecule absorbs two photons simultaneously, thereby producing the same excitation as a single photon of twice the energy. Thus the fluorophores requiring UV or visible excitation can be excited using NIR radiation that is less strongly attenuated by absorption and scattering – and is also less phototoxic. Because the efficiency of this nonlinear effect is proportional to the square of the intensity of the incident radiation, it is possible to arrange that the intensity of the beam is only sufficient for two-photon excitation to produce an appreciable fluorescence signal in the focal plane. Thus all detected photons can be assumed to have originated from the focal plane and optical sectioned (confocal-like) images can be obtained without any confocal aperture at the detector. In this way the longer wavelength excitation and increased photon collection efficiency permit multiphoton microscopy to image significantly deeper than confocal microscopy and imaging depths of up to ~500 mm are possible. If one uses an excitation wavelength at which there is little or no fundamental absorption in the sample, only that part of the sample in the focus of the beam will interact with the radiation and so photobleaching and photodamage damage are greatly reduced throughout the volume of the sample – unlike the situation for the confocal microscopy where the sample is excited throughout its volume even when photons from one specific depth are being detected to form a sectioned image. In practice, two-photon microscopy can permit extended 3-D imaging of many types of biological tissue without killing the cells under investigation and is widely applied to image live cells and tissue in vivo, although it is important to note that photobleaching is also a nonlinear process and increases with excitation intensity.
Other nonlinear laser scanning microscopy techniques have been developed that exploit second or third harmonic generation in the sample, e.g.2, 3. This approach is attractive because the intensity dependence of the nonlinearity means that the signal can be contrived to be generated only in the focal volume of the illuminating beam, providing the advantages of multiphoton microscopy, and for non-resonant nonlinear phenomena such as harmonic generation there is no energy deposited in the sample. Further nonlinear imaging modalities include Coherent anti-Stokes Raman scattering (CARS)4 and stimulated Raman scattering (SRS)5.
1 Denk, W., Strickler, J. and Webb, W., Science (New York, NY), 248 (1990) 73
2 Barad, Y., Eisenberg, H., Horowitz, M. and Silberberg, Y., Applied Physics Letters, 70 (1997) 922
3 Sun, C.-K.; Chu, S.-W.; Chen, S.-Y.; Tsai, T.-H.; Liu, T.-M.; Lin, C.-Y.; Tsai, H.-J.. Journal of Structural Biology 147 (2004) 19
4 Volkmer, A., Journal of Physics D: Applied Physics, 38 (2005) R59
5 Min W., Freudiger, C. W., Lu S.; Xie X. S., Annu. Rev. Anal. Chem. 62 (2011) 507
Wide-field optical sectioning microscopy
One drawback of laser scanning confocal microscopy (LSCM) and of multiphoton microscopy and other laser scanning nonlinear microscope techniques is that the sequential interrogation of the image pixels results in relatively long image acquisition times. These issues cannot be overcome by simply increasing the laser intensity since this causes unacceptable photobleaching and phototoxicity in the sample. The image acquisition rate can be increased without such penalties, however, by interrogating multiple pixels in parallel. This “tandem scanning confocal microscopy” was first implemented using a “spinning disc” scanner with multiple pinholes arranged to sweep across all pixels in the field of view1. The multiple pinholes serve as point sources for the illumination and as confocal apertures for detection, after which the images can be recorded using a wide-field detector. In the Photonics Group we use this spinning disc scanner to provide optical sectioning in FLIM microscopes and in automated FLIM multiwell plate platforms for high content analysis. Other approaches to parallelised pixel acquisition include splitting the excitation laser beam into multiple beams and scanning them in parallel. This has been implemented for both confocal and multiphoton2 fluorescence microscopy – the latter with the advantage that the emission does not need to be “descanned” since no (confocal) detection aperture is required. Alternatively, the sample can be illuminated with a line and the emitted or back-scattered light detected with a confocal slit aperture. The slit scanning ophthalmoscope, which is routinely used to image the retina, is an example of this approach.
An alternative approach to optical sectioning microscopy that utilises wide-field illumination and detection is that of structured illumination microscopy (SIM), for which the sample is irradiation with a spatially modulated intensity distribution. By acquiring multiple image acquisitions with different illumination patterns and applying appropriated computational algorithms to the images, it is possible to calculate optical sectioned images equivalent to confocal microscopy3 and even to increase the spatial resolution4 by a factor of 2 compared to wide-field microscopy. This provides a powerful tool for subcellular imaging that provides almost twice the resolution of a typical laser scanning confocal or multiphoton microscope in three dimensions. We are combining SIM with FLIM to enable cell signalling processes read out by FRET to be correlated with sub-100 nm morphological changes.
1 Petran, M., Hadravsky, M., Egger, M. D. and Galambos, R., Journal of the Optical Society of America, 58 (1968) 661
2 Bewersdorf, J.; Pick, R.; Hell, S. W., Optics Letters 23 (1998) 655
3 Neil, M. A. A., Juškaitis, R. and Wilson, T., Optics Letters, 22 (1997) 1905
4 Gustafsson, M. G., Journal of Microscopy, 198 (2000) 82
Light-sheet fluorescence microscopy
Recently there has been strong interest in light sheet fluorescence microscopy (LSFM), including techniques described as selective plane illumination microscopy (SPIM)1 and ultramicroscopy2 that realise optical sectioning via orthogonal illumination and imaging axes, with the sample being illuminated by a “sheet” of light, whose thickness defines the sectioning strength, and the excited sheet of sample fluorescence is imaged using a standard wide-field microscope, which defines the lateral resolution. This approach to optically sectioning fluorescence imaging can provide significantly reduced photobleaching and improved axial resolution compared to confocal microscopy. When configured with orthogonal excitation/imaging axes, the set-up is difficult to implement on a standard microscope frame since it requires separate excitation and imaging lenses, although some implementations that entail inclined but non-orthogonal excitation/imaging axes can be implemented using a single microscope objective on conventional inverted microscopes3 at the cost of reduced numerical aperture for the imaging channel. We have developed an approach to LSFM called oblique plane microscopy (OPM)4 that can be implemented on standard inverted or upright microscopes and is applicable to high content analysis with standard multiwell plates. Our configuration provides particularly fast 3-D imaging – realising > 600 optical sections/second or >25 volumes/second. LSFM is attracting particular interest because it provides a means to image mesoscopic samples up to mm in size with minimal phototoxicity. Mesoscopic imaging techniques are being rapidly developed and widely applied to imaging fixed and live biological samples and are providing new insights into structure and function of whole organisms. All optical techniques for imaging biological tissue, however, are challenged by the scattering of optical radiation and this ultimately limits their performance as the sample size increases.
1 Huisken, J., Swoger, J., Del Bene, F., Wittbrodt, J. and Stelzer, E. H. K., Science, 305 (2004) 1007
2 Dodt, H.-U., Leischner, U., Schierloh, A., Jahrling, N., Mauch, C. P., Deininger, K., Deussing, J. M., Eder, M., Zieglgansberger, W. and Becker, K., Nature Methods, 4 (2007) 331
3 Tokunaga, M., Imamoto, N. and Sakata-Sogawa, K., Nature Methods, 5 (2008) 159
4 Dunsby, C., Optics Express, 16 (2008) 20306
Total internal reflection fluorescence (TIRF) microscopy
It is possible to achieve sub-diffraction-limited optical sectioning with normal lateral fields of view at the high frame rates typically associated with wide-field imaging by using evanescent light to limit the excitation of fluorescence to a region very close to the coverslip using the technique of total internal reflection fluorescence (TIRF) microscopy. Note that TIRF microscopy only provides super resolution in the axial direction. As depicted in the figure, TIRF is implemented by arranging the excitation radiation to be incident at the coverslip at an angle larger than the critical angle for total internal reflection such that the light is internally reflected at the glass-sample interface with only an evanescent wave extending into the sample. Because this evanescent wave decays exponentially into the sample, typically only exciting fluorophores less than ~200 nm from the coverslip, TIRF microscopy has become widely used to image cell biology at or near the plasma membrane, e.g.1. The high selectivity of the evanescent wave excitation provides low background fluorescence with sufficient signal to noise to enable wide-field single molecule imaging near the coverslip if the sample is sparsely labelled, since this permits individual fluorophores to be visualised provided that they are separated by more than the lateral resolution of the microscope. This is an important step towards super resolved microscopy since the positions of individual fluorophores can be inferred by determining the centre of image of each molecule.
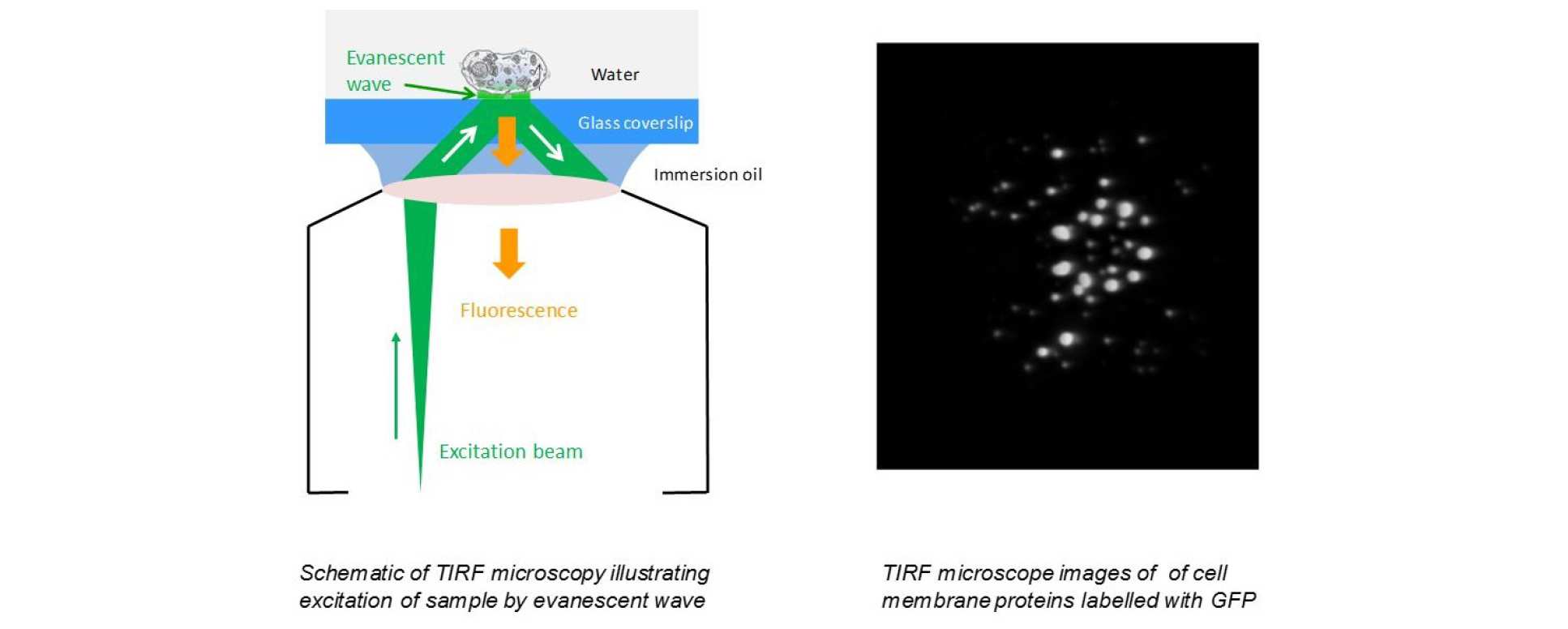
1 Axelrod, D., Traffic, 2 (2001) 764