
Dr Di Hu is a Research Associate in the Neurogenomics laboratories of Dr Nathan Skene and Dr Sarah Marzi at the UK DRI Centre at Imperial. She is developing single-cell multiomic sequencing approaches to study the human brain, and integrating sequencing at multiple genomic layers to investigate disease associated microglia in a humanised mouse model of Alzheimer's disease.

January 2021
Alexi Nott is a UK DRI Group Leader and Lecturer in the Department of Brain Sciences. He is investigating the epigenome of the human brain to understand how genetic variation contributes to age-related brain disorders.
Di: Thank you so much for doing this interview. First, I would like to understand where you're coming from and why you chose to go into epigenetics of the brain.
Alexi: Okay so I will start with my background. I did my PhD at UCL with Antonella Riccio where we were interested in looking at a chromatin modifier called HDAC2 (histone deacetylase 2). This enzyme is involved in removing acetyl groups from histones and is associated with transcriptional repression. We were looking to see how an external signal can lead to gene expression changes in the brain. The signalling pathway we were particularly interested in was mediated by nitric oxide. At the time, this was still a fairly new signalling molecule, particularly in neurons. We were able to show that when you stimulate neurons, you end up having an increase in nitric oxide. This leads to a post-translational modification on HDAC2, which then leads to changes in gene expression. My PhD studies was very focused on the role of this signalling pathway in the developing nervous system. After I finished my PhD I was interested in continuing along the line of looking at epigenetic regulation, but I wanted to explore it in a postnatal developmental setting. So I moved to MIT to work with Li-Huei Tsai and here we looked at the role of a different histone deacetylace HDAC3 and how it can regulate animal behaviour. I found that loss of HDAC3 in neurons in the brain led to behavioural changes that were similar to autism, and at the same time HDAC3 was shown to be a binding partner of MECP2. MECP2 is a very important protein implicated in Rett syndrome, which has autism-like features. I was able to tie a direct role of HDAC3 in Rett syndrome type behaviours and at a molecular level, I was able to show that MECP2 and HDAC3 in concert can positively regulate gene expression. This was at the time unexpected because they're both traditionally thought of as transcriptional repressors. The mechanism we showed was that HDAC3 is deacetylating a transcription factor that allows it to bind more strongly to DNA and elevate levels of gene expression. Having shown these mechanisms in mice, we were then able to recapitulate the molecular findings using induced pluripotent stem cells (iPSC), which were generated from a patient with Rett syndrome. We knew the Rett-causative mutation in this individual and we used CRISPR to correct it to the healthy version. We had isogenic lines, which are iPSC lines that are genetically identical apart from the Rett-causal mutation and we were able to recapitulate many of the molecular findings that we found in the mouse using the human iPSC approach. So, it’s translatable right?
Di: Yeah, I really like the mechanistic focus of this and how you found out what is happening at a molecular level.
Alexi: For me that was a very challenging project, but we went from animal behaviour down to mechanistically an aspect of what the proteins are doing. After that, because of the iPSC angle, I became more interested in looking at epigenetics in the context of the human brain. In addition, a lot of my previous work had been very focused on neurons, but the brain is a complex organ with many different cell types. A cell type that was becoming particularly prominent in the field of neuroscience when I was leaving MIT in 2016 was microglia. So I decided to do a second postdoc with Chris Glass at UCSD, who is an expert on tissue-resident macrophages. Here I was able to use genome-wide sequencing approaches to look at non-coding gene regulatory regions in different cell types of the human brain. To do this, we used a nuclei isolation approach where we are able to tag nuclei from specific cell types and isolate these using fluorescent activated nuclei sorting. Then with the cell type specific nuclei populations, we were able to do a number of different sequencing assays that allow us to define the non-coding gene regulatory regions of these different cell types, such as enhancers and promoters. From this we were able to incorporate genetic data from genome wide association studies (GWAS) that have identified genetic variants associated with an increased risk for different brain diseases. By incorporating the GWAS genetic data we were able to identify disease-risk cell types for different brain disorders. For example, we found Alzheimer's disease to be associated with enhancer elements that are active in microglia – the immune cells of the brain. Whereas for psychiatric disorders, like schizophrenia and autism, neuronal enhancers were associated with risk variants for these diseases. Because the risk variants for these different disorders are generally found located in non-coding regions, which means they are located outside of genes, we wanted to be able to tie these risk variants to their target genes. Quite often the way people think of non-coding gene regulatory regions or enhancers is that they affect the expression of the closest gene. However, we know that this is not really true, as sometimes they can affect genes that are more distal and sometimes they can affect the expression of multiple genes. To begin to get at this problem we have been collaborating with Bing Ren at UCSD. We did a chromatin conformation assay called PLAC-seq, or proximity ligation-assisted ChIP-seq, to tie disease non-coding risk variants to their target genes. This allowed us to identify new putative risk genes for Alzheimer’s disease. Then lastly, we validated one of the loci again using an iPSC approach where we took a cell type specific enhancer region that was identified in microglia and looked at the BIN1 locus, which is the second highest genetic risk locus for Alzheimer’s disease. We showed that when you delete this microglial specific enhancer in iPSCs and derive them into microglia, neurons and astrocytes, you only see an effect of the enhancer deletion on microglia. This confirms that the enhancer is really an enhancer and that it is cell type specific. Another interesting point of the study is that a lot of these risk genes are actually expressed in multiple cell types. BIN1 is expressed in all 4 cell types that we looked at, but the enhancer landscapes are quite distinct between each of the cell types, so by looking at the disease risk variants in the context of the enhancer landscape, you might be able to tease apart which cell type the genetic risk might be more relevant in.
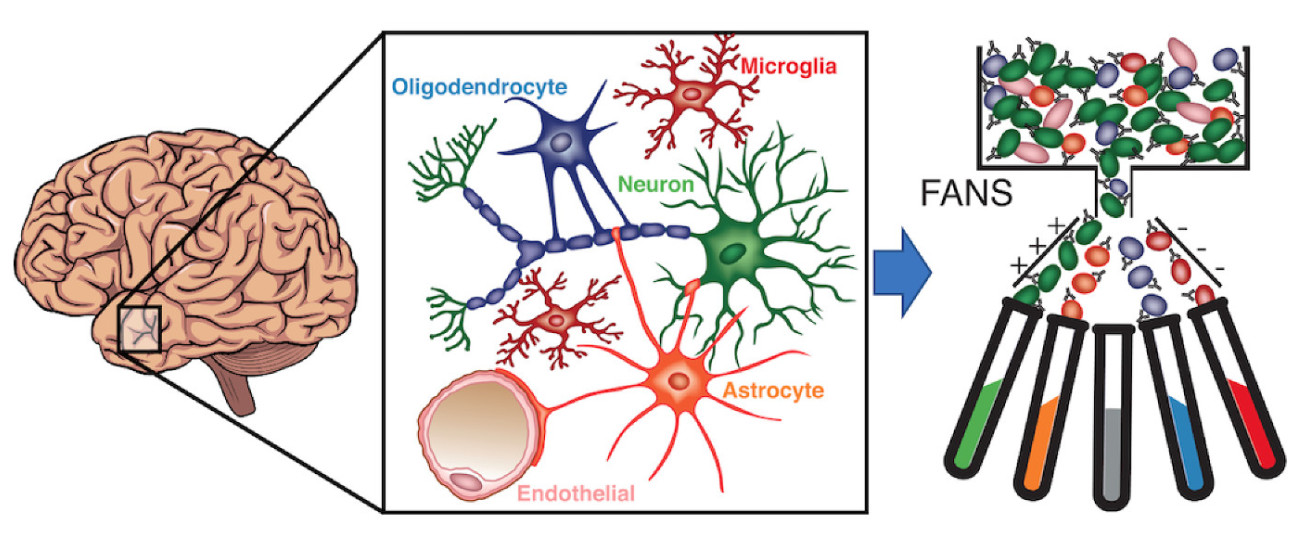
Di: Why did you focus on enhancers and not repressors?
Alexi: That’s a great question and it's true that just by looking at enhancer regions, we might be missing elements that are very important. This is something that we can do moving forwards. We can look at additional histone marks that will allow us to differentiate between repressed or silenced regions and also enhancers that are not necessarily active but are primed to be active in the future. There are ways of doing this by looking at different combinations of histone marks. Going forward, we can repeat similar experiments, but introduce more epigenetic marks.
Di: Do you think some of these epigenetic changes are purely due to the environment, or could there be some genetics underlying these epigenetic changes seen in dementia?
Alexi: I don't think we have a clear answer to that. There are studies that have begun to look at epigenetics in the context of disease. My original paper was looking at epigenetics of different cell types in a healthy context. There are other people who have looked at epigenetic changes associated with disease versus healthy ageing. To date, these are taken predominantly by an approach using bulk tissue, e.g., studies done by Dr Sarah Marzi and Dr Raffaella Nativio who are also in the department. The changes that they see from what I understand imply that there is not a perfect overlap between the genetic risk locations versus those that are actually changing in the context of disease. This could be exactly what you suggest that epigenetic changes due to the environment might be playing a role. What I believe is likely going to be the case is that it will be a combination of both – that the environment is going to cause epigenetic changes that are separate from genetic risk regions. It’s a hard question. When you’re looking for epigenetic changes between control versus disease, you’re looking at broad changes across the genome, and you’re comparing many different individuals where the genetics between those individuals are different. One thing that is more consistent between those individuals is probably changes due to the environment. That could be environmental risk factors such as ageing. It could also be the brain microenvironment. When you're looking at control versus disease, you're already looking at the disease at a later stage where there's going to be things like amyloid plaques and tau. In that cohort you're going to see environmental changes that are more likely to be in common, and potentially pick up epigenetic changes that are due to the environment. However, on a genetic level, the individuals are going to be quite heterogeneous, so I think it's going to be harder to tease out epigenetic changes that are directly driven by the genetics. It doesn't mean that the genetics isn't having an epigenetic influence on gene expression. I just think it will be harder to elucidate that by looking at control versus disease in a heterogeneous population of people.
Di: That is a really good point. How do you plan to study the impact of normal ageing on dementia and the role of environmental factors on dementia?
Alexi: Ageing is the biggest risk factor for Alzheimer’s disease. I want to do this using two separate approaches. One is to look at how epigenetic regions, the activity of them, change in different cell types across a lifespan. How do cell type specific epigenetic changes alter from developmental stages and early postnatal life through to adulthood and onto ageing? There’s a number of reasons why I think this might be important. For my current datasets I'm looking at enhancer regions that are active from paediatric epilepsy patients. The limitations of this cohort is that there might be gene regulatory regions that become more active later in life that could be relevant for disease that I'm missing from looking at such an early time point. On the flip side, because ageing is so important, another way to look at it is from the reverse. How early do some of these enhancer regions associated with risk factors become active? Even though you don't see anything clinically until later in life, at what point do these enhancer regions become biologically relevant in the context of disease? To tie in the environmental aspect to this, one environmental factor that might be important is early life infections. It has been shown that having an infection can lead to immune priming or immune tolerance. There's been a recent publication from Jonas Neher’s group that showed this can have an effect on the function of the immune system in the brain and could possibly contribute to the etiology of brain diseases. Taking the idea from this study, it would suggest that things that happen early in life could have epigenetic consequences that may affect you later in life. So knowing when some of these epigenetic risk regions become active and susceptible would be important. I think it's also important to know how epigenetic regions are changing in the context of disease, and then to be able to compare whether changes that are associated with ageing are distinct from those that occur in disease, or are they an exacerbation of normal ageing? It is also key to understand the changes occurring in different brain cell types. By looking at the epigenome you can infer transcriptional or gene regulatory programs that are being dysregulated and then in theory you might also be able to try and identify cell type specific therapeutic avenues by looking at these cell type specific changes. Okay and the second part of your question was to do with the environment, right?
Di: Yeah
Alexi: So for me the environment is interesting. While these brain disorders have been shown to have a heritable component, it has also become clear that as well as the genetics there are environmental factors that will dictate things like the onset of the disease, the rate of progression and the severity. The attractive thing about looking at environmental factors is that they are modifiable, so the idea here is that by just changing things associated with your lifestyle, you can potentially have an influence on disease progression and outcome. Environmental factors can have long lasting changes on cell function and this is likely to be captured at the level of the epigenome. I hope to look at different environmental risk factors that are associated with disease to see what effect they have on the epigenome of different cell types in the brain. I will start with looking at microglia because neuroinflammation is a hallmark of Alzheimer’s disease. The ultimate goal would be to integrate changes that are associated with the environment at the epigenome and see how these correlate with changes associated with ageing or disease. The hope here is to identify regions that are influenced by the environment that might be exacerbating or ameliorating changes associated with disease. The ultimate goal would be to try to provide an intervention for what might be going on in the disease context.
Di: Environmental factors contributing to disease would indeed be helpful to understand to inform lifestyle changes that could slow disease progression. With all the science you've done and plan to do, what is a lab culture that works well for you and how do you plan on implementing this?
Alexi: So for me lab culture is very important. I'm hoping to build on the examples that have been provided by my past mentors. For me, a critical aspect of this is for the lab to have a sense of a common goal or vision that they all feel a part of. It’s important for me to be able to communicate that and to build a sense of a team. I’m also hoping to extend that outside of my lab. There are a number of people within our department and also within the UK DRI as a whole that work on similar, but complimentary and synergistic problems to what I'm doing. The idea is we'll have a good team environment within the lab that will be part of an even bigger entity. Within our department this would include people like Dr Sarah Marzi, Dr Nathan Skene and Dr Raffaella Nativio. As a part of this, something that I'm hoping to encourage and promote is open communication. I want people to feel comfortable to express their ideas. I think this is important for the health of the lab as well as also for scientific innovation. I'm also hoping to have a very diverse lab. As someone who identifies as a gay scientist this has been key for me to feel comfortable in my own working environment. I believe this is important for the mental well-being of the lab members and it will also bring in new ideas and perspectives. In summary, the three main things are that there's a common goal and vision that's communicated well, open communication in the lab, and diversity.
Di: Very nice. What do you like most about science?
Alexi: For me it’s being able to explore the unknown and to see something for the first time that no one else has really seen before. There’s an excitement that you get from that. On top of this is the feeling that you're part of a team and then as you start to go to meetings and conferences, that you're part of a larger community. Then you really feel like you are contributing to this bigger picture, which is something that I think is well-promoted by the UK DRI. These are things that make me most excited about science.
![]()
