The first grant formalising the formation of our molecular systems engineering (MSE) area finished in 2013. At the Industrial Consortium Meeting of December 2013 the impressive achievements of the group were clear. In numbers, following the initial £3.6M awarded by the Engineering and Physical Sciences Research Council (EPSRC), we have secured £8M in complementary funding, have published over 50 papers and accumulated over 600 citations to our work in the duration of the grant. We had a very fruitful away day for the team in October 2013, to take stock on what has been achieved and to plan for an exciting and successful continuation of activities. We also have a new website: http://molecularsystemsengineering.org/, please visit it to see some of our recent papers and other work!
Our research continues, underpinned by a £1.8M Platform Grant (EPSRC) which supports our next generation of post-doctoral researchers, and with the support of BMS, Chemistry Innovation, GSK, P&G and Syngenta. The heading for this work is “Molecular Systems Engineering of High-Value Structured and Formulated Products”. We are heading to tackle key fundamental challenges for the prediction of fundamental properties of pharmacological and biologically relevant compounds. Our plan is to advance the tools of molecular systems engineering to tackle some of the key molecular challenges associated with improving the productivity of drug discovery and processing by improving our predictive capability of the relevant physical properties. We are collaborating with industry across several sectors, including pharmaceuticals (e.g., Pfizer, Novartis, Dynamic Extractions, GSK, BMS) and oil & gas (e.g., Shell, BP, Qatar Petroleum).
We are tackling these challenges by bridging the scales from the sub-atomic (quantum) to the macroscopic, and developing state-of-the-art techniques at the atomic and coarse-grained level, which lead to robust methods for property prediction and molecular design. Three of our key recent publications are highlighted:
Physical property prediction for the 21st century: the SAFT-γ Mie group contribution approach
Vasileios Papaioannou, Simon Dufal, Thomas Pogiatzis, Claire S. Adjiman, George Jackson, Erich A. Müller, Amparo Galindo
Thermodynamic methodologies are continuously being developed and improved in order to meet the industrial requirements for accurate property prediction in an ever-expanding range of applications. An important consideration or application within integrated process and solvent design is the predictive capability of the platform, commonly perceived as the ability of a method to predict the properties of a system for which no experimental data is available. An important class of predictive thermodynamic models are group contribution (GC) approaches, where molecules are modelled based on their corresponding chemical moieties. The SAFT- γ Mie group contribution approach brings together the molecular rigour, accuracy and versatility of the SAFT formalism with the predictive capabilities intrinsic of the GC concept.

An exciting application of such predictive approaches is in the description of the solubility of complex molecules, such as active pharmaceutical ingredients (APIs), in solvents and solvent
mixtures. The advantage of a predictive approach in this case is that the parameters required for the solubility prediction can be developed from experimental data for simple systems. Hence, the solubility of a given API is obtained in a fully predictive manner, without the requirement for API-specific experimental data for the parameterisation of the model. The potential of the SAFT-γ Mie approach in predicting the solubility of APIs in organic solvents is shown in Figure 1.
Sample publication: Papaioannou, V., Lafitte, T., Avendaño, C., Adjiman, C. S., Jackson, G., Müller, E. A. and Galindo, A., “Group contribution methodology based on the statistical associating fluid theory for heteronuclear molecules formed from Mie segments”, J. Chem. Phys. 140, (2014) 054107.
Accelerating chemical reactions by computer-aided molecular design.
Heiko Strübing, Zara Ganase, Eirini Siougkrou, Aikaterini Diamanti, Amparo Galindo, Claire Adjiman
What is the best solvent for a given chemical reaction? Given that the rate and selectivity of chemical reactions can vary by several orders of magnitude in different solvents, this question has important ramifications for the exploration of novel reaction routes and the development of industrial processes. We have been developing a methodology for optimal solvent design for enhanced reaction kinetics, QM-CAMD (cf. Figure 2), which relies on the integration of continuum solvation quantum mechanical calculations into a computer aided molecular design (CAMD) framework. This approach has been successfully applied to the SN2 reaction of phenacyl bromide and pyridine, leading to a 40% increase in the reaction rate. The results have been verified experimentally, using in-situ kinetic monitoring techniques.
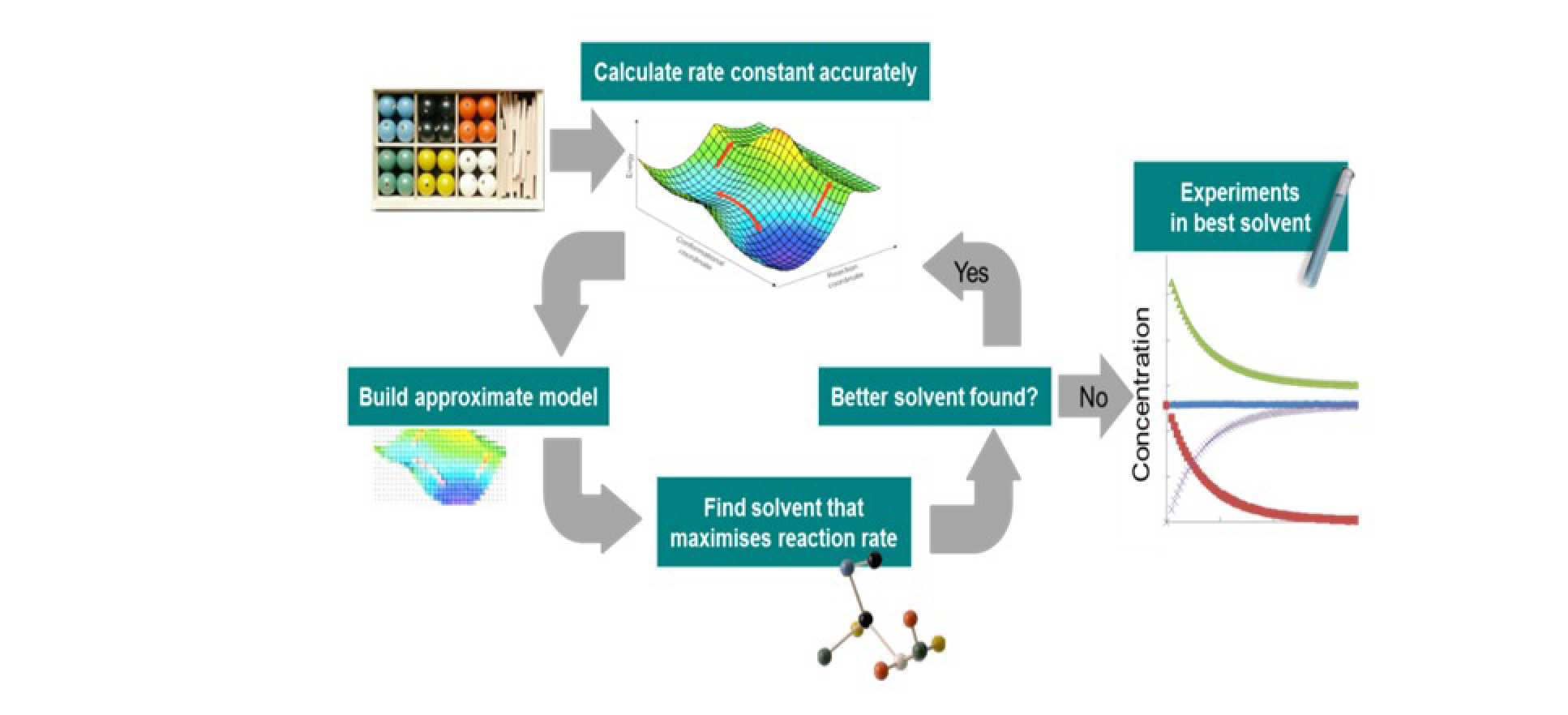
A central aspect of the proposed methodology is the use of a surrogate model. In initial work, we have used simple linear free energy relation, the solvatochromic equation. In more recent work, we have explored the use of more reliable surrogate models, in particular a kriging model, and found that it leads to further improvements in the solvents designed. On this basis, we have been extending the range of applicability of the approach by considering other reactions, including a Cope elimination reaction, in which the solvent can shift the equilibrium very significantly, and a Williamson reaction in which C-alkylation or O-alkylation products can be obtained preferentially depending on the solvent. We have also been further extending the methodology to include solubility and temperature effects on reaction kinetics.
Sample publication: Struebing, H, Ganase, Z., Karamertzanis, P.G., Siougkrou, E., Haycock, P. Piccione, P.M., Armstrong, A., Galindo, A., Adjiman, C.S., “Computer-aided molecular design of solvents for accelerated reaction kinetics”, Nature Chem. 5 (2013) 952-957. Highlighted in News and Views article, D.G. Truhlar, “Chemical Reactivity: Inverse Solvent Design”, Nature Chem. 5 (2013) 902-903.
Systematic methodologies for crystal structure predictions
Christina-Anna Gatsiou, Matthew Habgood, Isaac Sugden, Manolis Vasileiadis, Claire Adjiman, Costas Pantelides
Methodologies for the systematic prediction of the polymorphs of organic molecules solely from the knowledge of the molecular connectivity diagram have undergone rapid improvements in the last few years. Following the success of our CrystalPredictor/ CrystalOptimizer codes in identifying the correct structure for the largest molecule considered to date in a Blind Test (molecule XX), we have been making further improvements to our algorithms in order to increase the range of molecules which can be investigated (in particular, molecular size), and to increase the accuracy of the relative energy calculations. Major areas of development include (i) the implementation of improved local approximate models within CrystalPredictor, which makes it feasible to treat larger molecules reliably; (ii) the development of a methodology to improve the accuracy of lattice energy calculations and to embed existing experimental knowledge within the Crystal Structure Prediction methodology (iii) the development of a methodology for the rapid evaluation of free energy, enabling temperature effects to be taken into account. This latter development has been applied successfully to small molecules. For example, in the case of tetracyanoethylene, it has been shown to lead to the succesful prediction of reversal in stability of the cubic and monoclinic forms at higher temperatures, with the monoclinic form becoming more stable, as illustrated in Figure 3.
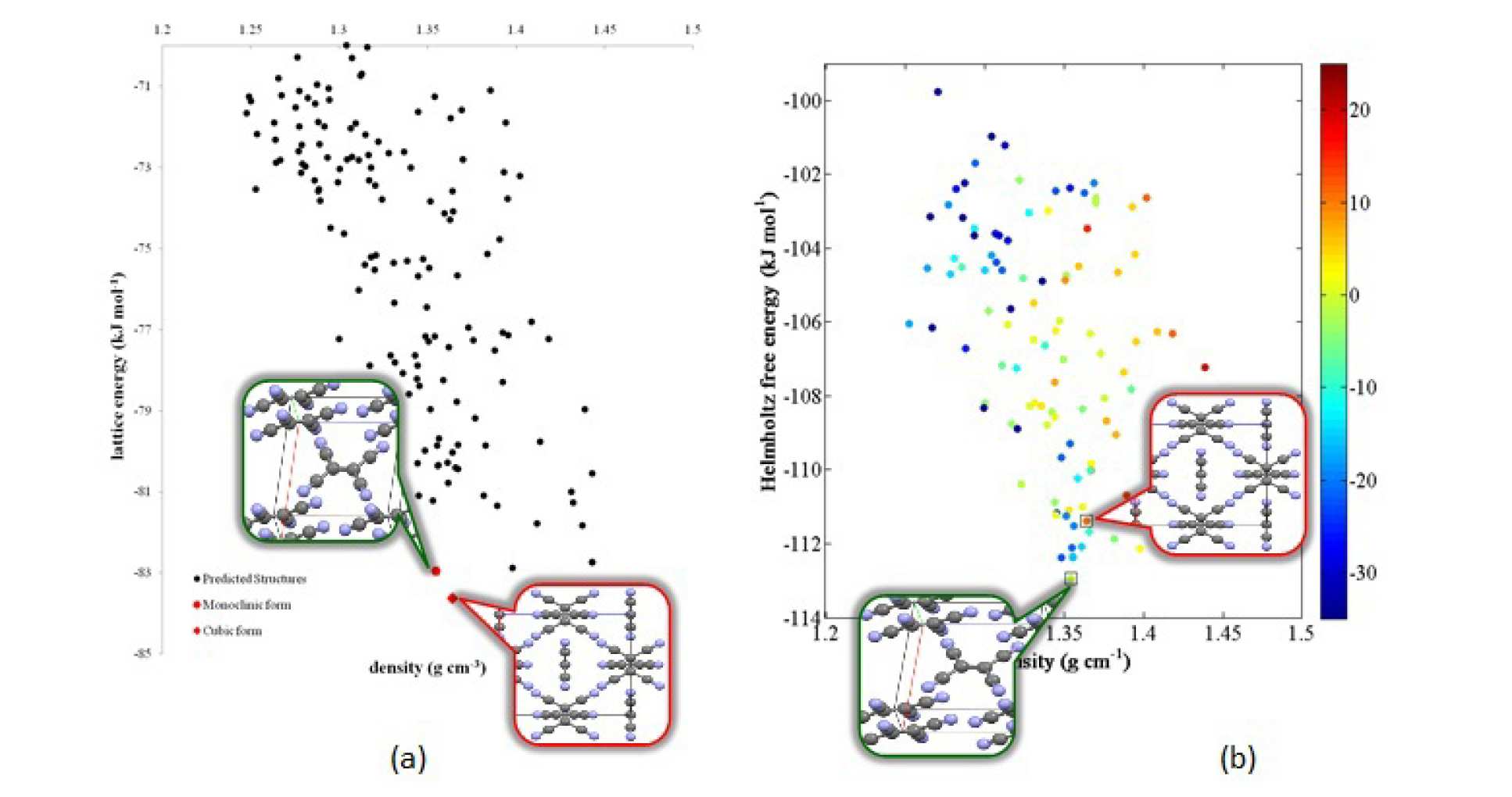
Sample publication: Pantelides, C.C., Adjiman, C.S., Kazantsev, A.V., “General computational algorithms for ab initio crystal structure prediction for organic molecules”, Top. Curr. Chem., Springer Berlin Heidelberg (2014).
![]()