Project submission process
1. Project discussion
Please get in touch with us at the earliest stage possible. Our knowledge and subsequent advice can enable your grant to go further and create better quality sequencing data.
Who to contact about your project:
Standard RNA-seq/DNA-seq - Nik Matthews, Katerina Rekopoulou and Benjamin Phillimore
Spatial Genomics - Nik Matthews and Nathalie Lambie
Proteomics or Single-Cell Genomics - Nik Matthews and Katerina Rekopoulou
2. Quotation
Sign and return the quotation received (ask for any changes needed before signing).
3. Sample submission form
Samples can only be submitted once ‘project tag’ has been given (this can be found on the quote). Complete the submission form and return it to us prior to submitting the samples to the Facility.
To avoid delays, it is important to adhere to the following instructions when filling in the submission form:
- Sample name to consist of ALPHA NUMERIC characters and hyphens only.
- NO OTHER PUNCTUATION MARKS OR SPACES to be included.
- Do not label your sample as a number eg ‘1’. Please label as ‘sample-1’ etc.
- Leave no empty rows.
- DO NOT forget to annotate the top section of the submission form, for example:
|
PrincipleInvestigator: |
David Williams |
|
MainContact: |
David Williams |
|
ProjectID: |
(leave empty) |
|
ProjectTag: |
williams_10-5-2023_mRNA |
|
QuotationNo: |
IGFQ001583 |
If you requested data analysis, we will need you to provide additional information regarding the experimental design, metadata and sample grouping. For this quote, we assume that each sample is only used once for data analysis, should any sample be needed more than once (e.g. control vs group1 and control vs group2) the cost for analysis will be adjusted by counting the control sample(s) twice.
4. Sample drop-off
- Prepare and deliver: samples/libraries to the facility in accordance with Sample Submission Guidelines (further information below). We can only proceed if the guidelines are followed exactly so please read them carefully.
5. Project starts
- Initial QC: Your samples/libraries/data will be initially quality checked (QC) to make sure that the quality is acceptable for the requested application.
Sample submission guidelines
- We require pools to be submitted in low bind 1.5mL Eppendorf tubes. The tube must be clearly labelled with the project tag from the quote, i.e. edwards_15-2-2021_ribodepletion. If the project tag has multiple pools then the sample tube must be also labelled with the pool number too, concordant with the submission form. Pool number in relation to samples should be stated in the submission form.
- Do not mix samples with UMI’s with samples without as this causes big problems when demultiplexing.
- Submissions for low numbers of reads (<1200 million) may suffer delays in sequencing due to maximising sequencing efficiency. Try and batch your samples together to achieve this.
- MINIMUM SUBMISSION (without arrangement with IGF): 100M reads
Indexing
Any misinformation regarding index sequences will invariably cause delay in data being created therefore it is vital that the submission form is filled in correctly and accurately.
- When supplying indexing information please quote the MiSeq indexes for i5 and i7. Do not provide primer sequences (P5 and P7).
- The index code should be entered under "index 1" (and / or index 2), "ID" and "Sequence" wells.
- Only add the index sequence to the sequence box on the submission form. Do not add anything else.
- It is recommended that ONLY dual index 8bp or dual index 10bp indexes are used. The use of 6bp single indexing is not recommended.
- The following box on the submission form should be left empty, unless using custom primers :

- Specifically, for 10x, we only require the index code under the index 1 sequence column; please do not include the index sequences. When submitting samples using 10x index indices, do not add any extra zero’s, e.g. SI-TT-A1 and not SI-TT-A01.
Pooling
-
- Prior to pooling barcoded libraries, it is essential to normalize the molar concentration of the libraries to ensure that an equal number of reads is generated for each library. Pre-made libraries are to be pooled by the submitter.
- Samples to be provided at highest possible molarity and volume. Absolute minimum for one sequencing run is 12µl @ 2.5nM.
1. RNA Extraction:
Total RNA can be extracted using common RNA-extraction kits such as Qiagen RNAeasy kits, Zymo kits (Direct-zol or Quick-RNA miniprep kits) or Invitrogen TRIzol. Note, RNA extraction must be carried out by the collaborator; we only accept extracted RNA samples.
2. DNase Treatment:
It's essential that the samples have been DNase treated as part of the extraction process in order to eliminate the background genomic DNA contamination. You should only use enzymatic DNA removal methods (TURBO DNase, ThermoFisher), we do not recommend column-based methods. Your RNA samples will be checked for genomic DNA contamination and if the contamination is found to be higher than 2%, an additional treatment will be done by us as part of the library preparation process.
3. RNA Quantity and Quality:
Quantity: A minimum of 15μL (more if you have enough or if it's low concentration) and a minimum amount of 300ng.
Quality: For mRNA applications, we require high quality RNA (minimum RIN of 7). For total RNA, we can accept lower RIN scores. We will check this as part of our internal QCs, so we will also ask for a second plate with 5μl of RNA, in order to minimise the free-thaw of the main sample plate.
4. Labelling:
Please use IGF identifiers only. Label the side of the plates and adhesive covers with the project tag found on the formal quote e.g. edwards_15-2-2021_ribodepletion. No other information is needed.
5. Sample Shipping
We only accept samples in 96-well PCR plates and won't accept samples in Eppendorf tubes or different types of plates (i.e. cell culture plates).
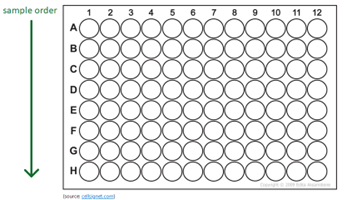
The samples should be aliquoted vertically; A1 to H1 then A2 to H2, etc, with no gaps between the samples.
Ensure your plates are covered with suitable plate sealers - qPCR optical covers are not recommended due to poor adhesion.
Dry ice shipment only.
Important:
Following the QC of your RNA samples, we will proceed with samples that meet the minimum requirements for the chosen kit. If any samples fails our QC, we'll contact you and give you the option to proceed, remove the sample(s) from the batch for library prep, or consider alternative library construction methods. The project and final invoice will be changed accordingly.
Sample submission guidelines.docx
1. DNA Extraction:
Genomic DNA should be purified using a column-based purification protocol such as the Qiagen DNeasy kit, or one of the DNA purification kits from Zymo Research (Quick-gDNA MiniPrep, Quick-gDNA Blood MiniPrep, or ZR Genomic DNA-Tissue MiniPrep). We recommend elution of the DNA in DNase-free water or tris buffer that does not have EDTA as it may interfere with enzyme activities during the library preparation. Extractions to be done by the submitter as only extracted DNA samples will be accepted.
2. DNA Quantity and Quality:
Quantity: A minimum of 15μL (more if you have enough or if it's low concentration) and a minimum amount of 300ng.
Quality: The minimum quality requirements (DIN score) specific to protocols.
3. Labelling:
Please use IGF identifiers only. Label the side of the plates and adhesive covers with the project tag found on the formal quote e.g. edwards_15-2-2021_ribodepletion. No other information is needed.
4. Sample Shipping
We only accept samples in 96-well PCR plates and won't accept samples in Eppendorf tubes or different types of plates (i.e. cell culture plates).
The samples should be aliquoted vertically; A1 to H1 then A2 to H2, etc, with no gaps between the samples.
Ensure your plates are covered with suitable plate sealers - qPCR optical covers are not recommended due to poor adhesion.
Dry/wet ice shipment.
Important:
Following the QC of your DNA samples, we will proceed with samples that meet the minimum requirements for the chosen kit. If any samples fail our QC, we'll contact you and give you the option to proceed, remove the sample(s) from the batch for library prep or consider alternative library construction methods. The project and final invoice will be changed accordingly.
